一种植物油中酚酸类化合物的测定方法与流程
本发明属于植物油的分类及鉴别技术领域,具体涉及一种植物油中酚酸类化合物的测定方法。
背景技术:
酚类化合物包括简单酚、多酚和酚酸,是植物重要的次生代谢产物。目前,酚类化合物已达4000余种,且分为12个亚类。酚酸类成分作为酚类化合物重要的一类,由于其抗氧化活性,癌症预防、抗炎以及与改善心血管疾病等作用密切相关而备受关注。不同植物油含有不同种类及含量的酚酸类化合物,它们是植物油微量组分中重要的一部分,不仅有助于植物油对氧化的稳定性,而且能够影响其感官特性和营养质量。因此,针对微量酚酸类化合物建立一种灵敏、快速的检测方法显得尤为重要。
目前,酚酸类化合物的检测方法研究虽备受关注,但其检测主要集中在酚酸类化合物含量较高的葡萄酒、中成药以及粮食中。
例如《超高效液相色谱法快速测定葡萄酒中酚酸》(唐柯,马磊,徐岩,等.中国酿造,2015,34(12):157-161)和《葡萄酒中11种酚酸的反相高效液相色谱测定方法研究》(陈建业,温鹏飞,战吉成,等.中国食品学报,2006,6(6):133-138)中即公开了葡萄酒中酚酸类化合物的检测方法。
再例如《高效液相色谱法同时测定款冬花中四种酚酸》(冯海燕,杨晓辉,岳红坤,等.分析科学学报,2015,31(2):000281-284)、《银杏叶中银杏酚酸类成分含量测定方法研究》(王荞薇,谢媛媛,王义明,等.中国药学杂志,2015,50(2):167-173)中即分别公开了中药植物原料中的酚酸类化合物的检测方法。
再例如《超高效液相色谱法分析稻米酚酸化合物组分及其含量》(张娜,王国祥,abacarjosedaniel,等.中国农业科学,2015,48(9):1718-1726)和《固相萃取-高效液相色谱测定化感水稻根系分泌的酚酸类化合物》(郑新宇,叶仁杰,何印波,等.云南大学学报(自然科学版),2013,35(2):219-224)中即公开了粮食中的酚酸类化合物的检测方法。
相比于葡萄酒、中成药以及粮食,因植物油复杂的基质和较低的酚酸类化合物含量,植物油中酚酸类化合物的检测难度非常高,因此针对植物油中酚酸类化合物的检测鲜有报道。
酚酸类化合物常见的提取方法有液液萃取法、固相萃取法和超声辅助萃取法等。用于酚酸类化合物的分离检测方法多为高效液相色谱法、毛细管电泳电化学法和高效液相色谱串联质谱法。对于质谱法,因为质谱昂贵的价格限制了国内使用的普遍性,而高效液相色谱法和毛细管电泳电化学法多存在分析时间长,灵敏度不高以及前处理繁杂,特别是样品中的杂质对目标物的干扰较大,定量不准确,不能用于植物油中微量酚酸类化合物的同时测定。
技术实现要素:
为了解决现有技术存在的上述问题,本发明目的在于对针对植物油中基质复杂、酚酸类化合物含量低的特殊情形,针对植物油中的酚酸类化合物的检测方法进行深入研究。以期提供一种具有良好的线性范围及检测限,日内和日间精密度,回收率、重复性及稳定性较好,简单实用的植物油中酚酸类化合物的测定方法。
本发明为构建植物油酚酸类化合物的稳定方法学,构思采用spe-uhplc-dad法对植物油中的9种酚酸化合物进行检测分析,通过优化spe前处理以及超高效液相色谱分离参数,力求达到前述研究目的,建立一种快速、灵敏的检测方法。
通过深入研究,本发明揭示以下技术方案:
一种植物油中酚酸类化合物的测定方法,主要包括步骤植物油样品前处理和uhplc-dad色谱分析;
所述植物油样品前处理步骤中,以正己烷溶解植物油样品,再使用二醇基固相萃取柱(diol-spe)进行净化处理;
所述uhplc-dad色谱分析中,进行梯度洗脱,检测波长为280nm。
在以上方案的基础上,根据本发明的具体实施例。以正己烷溶解植物油样品中:每克植物油样品中加入2ml正己烷进行涡旋得到样品提取液。
在以上方案的基础上,根据本发明的具体实施例。使用二醇基固相萃取柱进行净化处理中:先依次用体积比为1:1的甲醇-正己烷混液对固相萃取柱进行活化,流速控制在2.0ml/min;活化完成后,将样品提取液以1.0ml/min流速过diol固相萃取柱,再用正己烷淋洗萃取柱,弃去全部的流出液,最后用甲醇或甲醇水溶液洗脱,收集全部的洗脱液,氮气吹近干,残渣溶于体积比6:4的甲醇-水溶液中,涡旋振荡1.0min,4℃下10000r/min离心5min,取上清液。
在以上方案的基础上,根据本发明的具体实施例。所述uhplc-dad色谱分析中,先以0.1%甲酸甲醇溶液作为流动相a进行梯度洗脱,再以0.1%甲酸水溶液作为流动相b进行梯度洗脱;流速0.3ml/min;柱温40℃;进样量2.0μl。
本发明的有益效果为:
通过构思spe-uhplc-dad法对植物油中的9种酚酸化合物进行检测分析研究,本发明获得的植物油中酚酸类化合物的测定方法快速、灵敏、实用性强。通过对16批植物油进行分析以及对比,不仅能够为植物油中微量的酚酸类化合物进行较为准确的定量研究,而且为植物油的分类及真假判定提供了理论基础。
在以上的基础上,本发明对所提供的植物油中酚酸类化合物的测定方法在线性范围、检测限、日内和日间精密度、回收率、重复性及稳定性等各方面进行了进一步深入研究评析,均具有良好的效果。对于植物油9种酚酸类化合物的检测结果最好,该检测方法的回收率在91.35%-103.21%之间,日内及日间精密度的rsd范围分别在0.3%~0.9%和0.6%~1.1%之间。
附图说明
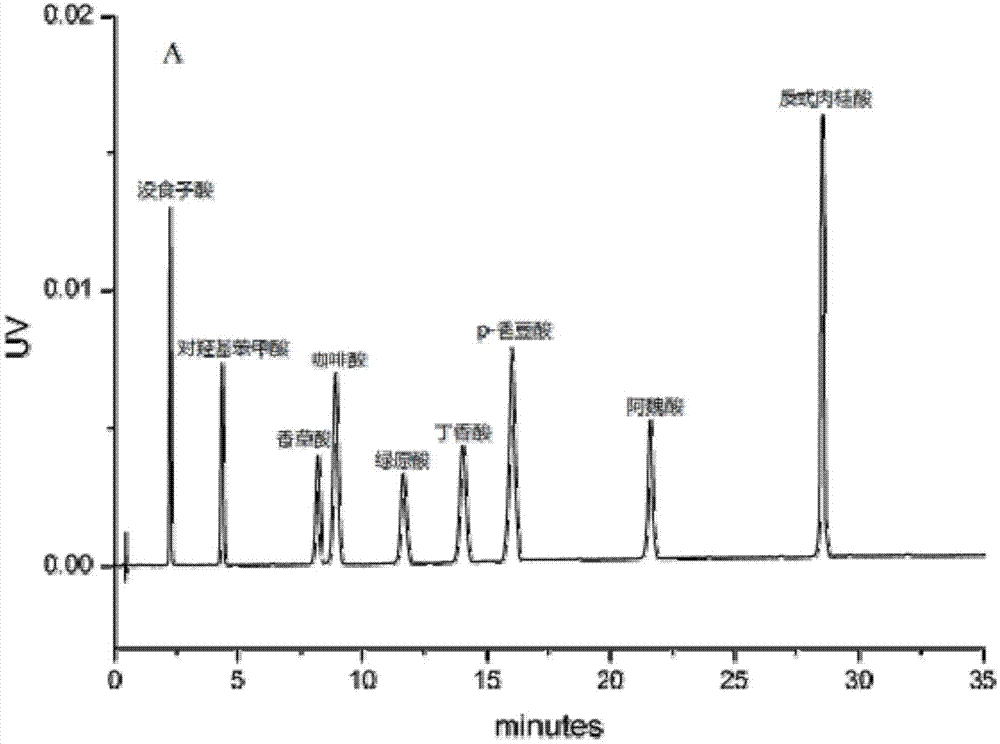
图1为甲醇-水体系(a)下9种酚酸类化合物的分离色谱图;
图2为乙腈-水体系(b)下9种酚酸类化合物的分离色谱图;
图3为不同流速的分离色谱图;图3中峰注:1.没食子酸;2.对羟基苯甲酸;3.香草酸;4.咖啡酸;5.绿原酸;6.丁香酸;7.p-香豆酸;8.阿魏酸;9.反式肉桂酸;
图4为不同温度的分离色谱图;图4中峰注:1.没食子酸;2.对羟基苯甲酸;3.香草酸;4.咖啡酸;5.绿原酸;6.丁香酸;7.p-香豆酸;8.阿魏酸;9.反式肉桂酸;
图5~8为部分植物油样品色谱图。
具体实施方式
下面结合具体实施例对本发明做进一步阐释。
一、在spe-uhplc-dad法的技术构思下,本发明提供一种植物油中酚酸类化合物的测定方法的具体实施例如下:
样品提取:取5.00g植物油样品于50ml聚乙烯管中,加入10ml正己烷溶液,涡旋1.0min,待净化。
样品净化:首先依次用10.0ml甲醇和10.0ml正己烷对固相萃取柱进行活化,流速控制在2.0ml/min;活化完成后,将样品提取液以1.0ml/min流速过diol(二醇基)固相萃取柱,然后再用10.0ml正己烷淋洗萃取柱,弃去全部的流出液,最后用10.0ml甲醇洗脱,收集全部的洗脱液,氮气吹近干,残渣溶于2.0ml甲醇-水溶液(6:4,v/v)中,涡旋振荡1.0min,4℃下10000r/min离心5min,取上清液,待测。
waters公司beh柱(50mm×2.1mm,1.7μm);流动相a为0.1%甲酸甲醇(v/v)溶液,b为0.1%甲酸水(v/v)溶液;采用梯度洗脱程序(见表1);流速0.3ml/min;柱温40℃;进样量2.0μl。检测波长为280nm。
表1梯度洗脱条件
二、在spe-uhplc-dad法的技术构思下,本发明实施例中还对流动相、流速、色谱柱温度的各项参数选择进入了深入研究,具体如下:
材料与试剂
15种植物油样品,包括初榨橄榄油8种(o1~o8)、菜籽油3种(c1,c2,c3)、以及亚麻油2种(y1,y2)均购自本地区大型超市,精炼橄榄油3种(j1,j2,j3),由实验室自制。
标准品没食子酸、对羟基苯甲酸、香草酸、咖啡酸、绿原酸、丁香酸、p-香豆酸、阿魏酸和反式肉桂酸;甲醇和乙腈为色谱纯;其他试剂均为分析纯。
仪器与设备
ultra-performance超高效液相色谱仪,配有二极管阵列检测器(dad);sartoriusbp211d型电子分析天平(十万分之一);bondelutdiol固相萃取柱;kq3200db数控超声波清洗器。
标准品溶液的制备
分别精密称取没食子酸、对羟基苯甲酸、香草酸、咖啡酸、绿原酸、丁香酸、p-香豆酸、阿魏酸和反式肉桂酸标准品10.00mg,置10ml棕色量瓶中,加甲醇适量溶解并稀释至刻度制成1.0mg/ml的混合标准储备液,4℃保存。分别精密量取混合标准储备液取1ml,氮气吹近干后,流动相配成0.10、0.25、0.50、1.00、5.00、10.00、50.00、100.00μg/ml的系列标准工作液,4℃保存,备用。
1)流动相的选择
流动相的组成体系是影响目标物分离及色谱峰形的重要因素之一,根据酚酸类化合物的极性,本实验在相同洗脱条件下(见表1),研究水-乙腈体系以及水-甲醇体系下9种酚酸类化合物的分离效果。根据目标物的保留时间、峰形以及分离度等因素确定最佳的流动相体系。如图1所示,当流动相为甲醇-水体系时,9种酚酸类化合物的分离度良好,且分离时间适中。如图2所示,当流动相为乙腈-水体系时,由于在反相色谱分离中,乙腈对于目标物的洗脱能力强于甲醇,在相同洗脱梯度条件下,9种酚酸类化合物的保留时间均有提前,且咖啡酸和香豆酸完全重合,未能达到分离要求,鉴于此,本发明选用甲醇-水体系为检测酚酸类化合物的最佳流动相体系。
2)流速的选择
在传统反相液相色谱分离过程中,在系统压力允许的范围内,流速的大小与整个分离分析时间的长短有着必然的关系,当流动相的流速越大,被分离物质出峰时间越快,峰型尖锐,灵敏度较高;较小的流速会造成分析时间过长,色谱峰展宽而影响检测的灵敏度。本实验使用超高效液相色谱,考虑到流动相的黏度和系统最高压力的限制,流速在0.1~0.4ml/min范围内进行优化,见图3;当流速为0.1ml/min时,在固定的35min分离时间内,反式肉桂酸(9)因为较强的保留而未被洗脱下来。此外,香草酸(3)、咖啡酸(4)和绿原酸(5)拖尾严重,没食子酸(1)甚至出现了峰分叉现象,不仅不能满足快速、高通量分析,而且还影响定量的准确性及分析的灵敏度。当流速为0.4ml/min以上时,由于梯度洗脱过程中随着时间的增大,当甲醇比例在达到30%时,系统压力超出了最高压力的85%,为了保证较好的灵敏度、系统压力以及尽可能与杂质的分离,本发明最佳流速选择为0.3ml/min。
3)色谱柱温度的选择
温度对目标物在反相色谱中分离的影响主要体现在主要体现在柱温的改变,直接影响到流动相的密度及黏度以及样品分子的传质速率,从而改变目标物与固定相和流动相之间的作用力,影响其保留时间;为了使9种酚酸类化合物得到较好的分离,本实验在保持其它色谱条件不变的前提下,在25~40℃的范围内考察了柱温对目标物分离的影响,见图4。结果表明,随着温度的升高,酚酸类物质的保留时间逐渐减小。当温度为35℃时,目标物中香草酸及咖啡酸分离度(r)仅为1.1,不仅不能满足分离要求,在实际样品的应用中使得杂质很容易干扰目标物,影响定量的准确性,当温度为40℃时,因为没食子酸与流动相及固定相之间作用力的改变,使得色谱峰出现了分叉,其余化合物的峰展宽增加,且咖啡酸和香草酸完全重合。当温度为25和30℃时,目标物均得到分离,为了保证9种酚酸类物质在较短时间内达到分离,并且在分析过程中相互之间不被样品中的杂质所干扰,本实验选择最佳分离温度为30℃。
4)固相萃取柱的选择
由于固相萃取柱操作简便、成本较低、能有效分离杂质和目标物,净化及富集效果明显优于液液萃取,鉴于此,本研究以9种酚酸类化合物的标准工作液(1.00μg/ml,10ml)作为目标物,分别采用hlb以及diol(二醇基)固相萃取柱通过上样及淋洗两个步骤考察9种酚酸类化合物的吸附效果,结果表明,当使用hlb固相萃取柱时,没食子酸在上样过程中已损失近70%;当使用diol固相萃取柱时,9种酚酸类化合物均有较好的吸附,使用正己烷进行淋洗也未检测到没食子酸以及其它目标物。因此,本实验确定diol为最佳固相萃取柱。
三、在以上的基础上,本发明所提供的实施例还进行了对本发明所提供的方法学的方法学考察,具体如下:
1)稳定性实验
取植物油样品(o1)5.00g,准确加入高、中、低三种不同浓度的9种被测组分混合标准工作液(0.25、1.00、5.00mg/l)各1.0ml,按照前述本发明所提供方法处理样品,按照前述本发明所提供方法中选择的所述色谱条件,分别在0、4、12、24、48h分别进样分析,每个时间点进样3次,取平均值。植物油中9种酚酸类物质(没食子酸、对羟基苯甲酸、香草酸、咖啡酸、绿原酸、丁香酸、p-香豆酸、阿魏酸和反式肉桂酸)的峰面积rsd分别为0.03%,0.48%、0.57%、0.52%、0.68%、0.69%、0.71%、0.65%和0.84%;结果表明样品溶液在48小时内稳定。
2)精密度试验
分别对仪器精密度进行考察;按照前述本发明所提供方法中所述色谱条件,准确加入高、中、低三种不同浓度的9种(没食子酸、对羟基苯甲酸、香草酸、咖啡酸、绿原酸、丁香酸、p-香豆酸、阿魏酸和反式肉桂酸)被测组分混合标准工作液(0.25、1.00、5.00mg/l)各1.0ml,重复进样6次,见表2;由表2可得9种目标物的日内精密度的rsd范围在0.3%~0.9%之间;日间精密度的rsd范围在0.6%~1.1%之间,由此可以看出该仪器对9种酚酸类物质具有较好的日间和日内精密度,能够满足植物油样品中9种主要酚酸类物质的检测要求。
表29种酚酸类化合物的稳定性及精密度
3)专属性实验
取所得植物油样品溶液(o1)、混合标准溶液、阴性供试品溶液,按前述本发明所提供方法中所述的色谱条件进样分析,将阴性样品与混合标准溶液色谱图和样品色谱图比较,在9种酚酸类化合物(没食子酸、对羟基苯甲酸、香草酸、咖啡酸、绿原酸、丁香酸、p-香豆酸、阿魏酸和反式肉桂酸)的出峰位置无干扰,表明该方法测定方法专属性好。
4)线性范围及定量限
分别精密量取混合标准储备液取1.0ml,氮气吹近干后,使用流动相配成0.10、0.25、0.50、1.00、5.00、10.00、50.00、100.00μg/ml的系列标准工作液,按前述本发明所提供方法中所优化的色谱条件进样(n=3),以峰面积平均值a为纵坐标,浓度c(μg/ml)为横坐标,绘制标准曲线;此外,将标准工作液(0.10μg/ml)逐级稀释,通过信噪比(s/n)为10考察定量限,(s/n)为3考察检出限。结果见表3,9个成分的相关系数r2均大于0.99,表明各成分在线性范围内线性良好。
表39种酚酸类物质线性范围及定量限
5)加标回收率试验
准确称取6份同一批(o1)植物油样品,准确加入高、中、低三种不同浓度的9种被测组分混合标准工作液(0.25、1.00、5.00mg/l)各1.0ml,按本发明所提供方法进行样品处理,按本发明所提供方法中所述的色谱条件进样分析,加样回收率实验显示,没食子酸、对羟基苯甲酸、香草酸、咖啡酸、绿原酸、丁香酸、p-香豆酸、阿魏酸和反式肉桂酸含量的加样平均回收率范围分别在91.35%~103.21%之间,植物油中9种酚酸类物质的回收率的rsd范围在0.36%~1.11%之间,结果见表4,结果表明本实验方法准确可靠。
表4植物油样品中9种酚酸类化合物的回收率
n.d:未检出
6)重复性试验
准确称取同一植物油样品(o1)6份,添加混合标准工作液(1.00mg/l)1.0ml,按本发明所提供方法进行样品处理,按本发明所提供方法中所述的色谱条件进样分析,并计算9种酚酸类化合物的含量及rsd,其结果可以看出,6份同一植物油样品(o1)中没食子酸、对羟基苯甲酸、香草酸、咖啡酸、绿原酸、丁香酸、p-香豆酸、阿魏酸和反式肉桂酸含量的rsd分别为0.8%、1.1%、0.9%、1.3%、1.2%、1.0%、1.2%、1.4%和1.6%。表明方法重复性较好。
四、在以上的基础上,运用本发明的实施例所提供的检测方法进行所收集的样品的测定,具体如下:
取所收集的17批次植物油样品,准确称取5.00g,按述本发明所提供方法进行样品处理,按前述本发明实施例所提供方法中选定的所述的色谱条件进样分析,每个样品重复进样3次,计算9种酚酸类化合物的含量,结果见表5;部分植物油样品色谱图见图5~8。
表516批植物油样品的含量(n=3)
a):1.没食子酸;2.对羟基苯甲酸;3.香草酸;4.咖啡酸;5.绿原酸;6.丁香酸;7.p-香豆酸;8.阿魏酸;9.反式肉桂酸
n.d:未检出n.q:未定量
由表5可以看出,反式肉桂酸在植物油中分布最为广泛且含量较高,在2.01-0.29mg/kg之间;其次,没食子酸为菜籽油中主要酚酸的类化合物,并含有微量的香草酸、咖啡酸以及反式肉桂酸;亚麻酸中酚酸类化合物的种类较多,主要包括没食子酸、对羟基苯甲酸、绿原酸、p-香豆酸以及反式肉桂酸,但它们的含量较少,仅在0.43~0.83mg/kg之间;橄榄油中含有较高含量的反式肉桂酸、阿魏酸以及咖啡酸,仅含有微量的没食子酸,与godoy-caballerom.p等(godoy-caballeromp,acedo-valenzuelami,galeano-díazt.simplequantificationofphenoliccompoundspresentintheminorfractionofvirginoliveoilbylc–dad–fld[j].talanta,2012,101(22):479-487.)的研究结论基本一致;值得注意的是,精炼橄榄油中除微量反式肉桂酸外,不含其它任何酚酸类化合物,这为判定精炼橄榄油及初榨橄榄油提供了一定的研究思路。
基于以上具体实施方式,本发明考察了植物油样品中9种酚酸类化合物的提取净化方式、流动相组成、流动相流速以及色谱柱温度对9种酚酸类化合物检测结果的影响。以正己烷溶解植物油后,使用二醇基固相萃取柱(diol-spe)对其进行净化,流动相为甲醇-水溶液,梯度洗脱;流速为0.3ml/min,检测波长为280nm;uplc-dad对9种酚酸类化合物的分离检测效果最好。该方法在测定植物油样品中的酚酸类时,具有良好的线性范围及检测限,且方法的日内和日间精密度,回收率、重复性及稳定性较好,简单实用。通过对16批植物油进行分析以及对比,不仅为植物油中微量的酚酸类化合物进行较为准确的定量研究,而且为植物油的分类及真假判定提供了理论基础。
本发明不局限于上述可选的实施方式,任何人在本发明的启示下都可得出其他各种形式的产品。上述具体实施方式不应理解成对本发明的保护范围的限制,本发明的保护范围应当以权利要求书中界定的为准,并且说明书可以用于解释权利要求书。
- 还没有人留言评论。精彩留言会获得点赞!