稀土掺杂磷酸钙荧光纳米颗粒在生物体内的定量检测方法与流程
本发明涉及生物检测技术领域,具体涉及一种稀土掺杂磷酸钙荧光纳米颗粒在生物体内的定量检测方法。该方法可用于研究荧光纳米颗粒在生物体内的分布与代谢过程。
背景技术:
磷酸钙是人体硬组织(骨头、牙齿等)的主要组成部分,其主要包括无定形磷酸钙(acp)、二水磷酸氢钙(dcpd)、磷酸八钙(ocp)、羟基磷灰石(hap)等。磷酸钙具有良好的生物相容性和生物活性,在生物医学领域已得到广泛应用。纳米磷酸钙更是由于较小尺寸、高比表面积、高吸附能力、高生物活性、高成骨能力等优点,在药物载体、生物复合材料无机相、纳米生物陶瓷等方面展现出良好的应用潜力。此外,考虑到稀土元素的荧光性、磁性,稀土掺杂磷酸钙纳米颗粒有望成为一种生物相容、可降解的荧光或磁显影剂,用于疾病的诊断。作为一种生物成像剂,稀土掺杂磷酸钙在生物体组织内的分布及代谢情况是必需研究的内容。
目前,可用于磷酸钙纳米颗粒体内示踪研究的技术主要有放射性同位素标记法和高分子荧光物质标记法。放射性同位素标记法具有检测灵敏度高等优点,但是该方法需要在较为严苛的专业条件下进行,并且放射性同位素对生物体有潜在的安全风险。高分子荧光物质标记法一般通过物理吸附或化学键合使高分子荧光物质附着在磷酸钙纳米颗粒的表面,在初期可以起到较好的示踪效果,但是随着磷酸钙纳米颗粒的溶解,高分子荧光物质会与磷酸钙分离而失去标记效果,因而无法起到长期的体内示踪作用。
发明人团队较早前公开了一种稀土掺杂磷酸钙荧光纳米粒子(re-ncap)的制备方法(cn105018086a)以及一种用于细胞内羟基磷灰石(hap)纳米粒子的定量检测示踪方法(cn103822906a),该检测示踪方法虽然能够准确的反应羟基磷灰石纳米粒子在细胞内的溶解情况并对其进行定量检测,但是由于没有设置空白对照组进行显著性差异分析,也没有计算稀土元素在生物组织中的提取率,因而准确性有待加强。此外,该检测示踪方法不能准确定量的检测出稀土掺杂磷酸钙在动物组织(包括硬组织、软组织及血液)和代谢物(尿液和粪便)中的含量。
在此基础上,发明人团队经过不断深入研究,开发了一种新的定量检测方法,该方法的检测极限浓度可达0.1nm,能够精确检测出稀土掺杂磷酸钙纳米颗粒在生物体内(包括硬组织、软组织及血液中)以及生物代谢物中的含量,还可以为纳米颗粒在生物体的分布代谢等研究提供一种精确的检测方法。
技术实现要素:
本发明的目的在于提供一种稀土掺杂磷酸钙荧光纳米颗粒在生物体内的定量检测方法,该方法包括以下步骤:
(a)在荧光增强液体系中建立稀土离子re的荧光强度-浓度标准曲线;
(b)利用酸溶液分别处理待测组生物组织样品、不含re-ncap的空白对照组生物组织样品,得到待测组匀浆和空白对照组匀浆;取适量待测组匀浆、空白对照组匀浆离心分离得到上清液,用荧光增强液稀释上清液后检测其荧光强度,并对待测组和空白对照组的单位质量或体积荧光强度值进行显著性差异分析。若两者之间不具有显著性差异(p≥0.05),说明待测组生物组织样品中不含re-ncap(即含量为0),到此结束不再进行后续操作;若两者之间具有显著性差异(p<0.05),则按照步骤(c)和步骤(d)的操作进一步确定待测组生物组织样品中re-ncap的含量。
(c)将re-ncap分别加入到相同体积的空白对照组匀浆和酸溶液中,离心分离得到上清液,用荧光增强液稀释上清液后检测其荧光强度,根据空白对照组匀浆和酸溶液的荧光强度计算单位重量或体积组织滞留率,进而根据待测组生物组织的重量或者体积计算出待测组组织提取率;
(d)综合考虑荧光强度-浓度标准曲线、组织提取率、匀浆体积、单位质量或体积荧光强度值、稀释倍数以及磷酸钙中稀土元素的摩尔含量,最终计算出待测组生物组织样品中re-ncap的含量。
按照上述方案,步骤(a)中荧光强度y与浓度x标准曲线的建立方法具体如下:以荧光增强液为溶剂,配制一系列不同浓度(7nmol/l以内)的稀土离子标准溶液,利用荧光分光光度计测定稀土离子标准溶液在特定激发波长(330-350nm,优选为340nm)下特定发射波长(如eu3+为618nm或者tb3+为488nm)的荧光强度,直线拟合得到荧光强度y与浓度x的关系为:y=b+nx。其中,b为截距,单位cps;n为斜率,单位cps/(nmol/l)。
按照上述方案,步骤(b)具体过程为:分别精确测量待测组生物组织样品和不含eu-ncap的空白对照组生物组织样品的质量或者体积,再加入酸溶液,得到待测组匀浆和空白对照组匀浆;取等量待测组匀浆和空白对照组匀浆,分别离心分离得到上清液,将上清液与酸溶液按照一定比例(体积比1:19)混合后得到上清稀释液,将上清稀释液与荧光增强液按照一定比例(体积比1:199)混合后得到混合液,在特定激发波长下测定特定发射波长的荧光强度。待测组和空白对照组的荧光强度值分别记为y1和y0,按照下述公式计算单位质量或体积组织荧光强度值(t):
其中,t为单位质量或者体积组织荧光强度值(单位,cps/g或者cps/ml);y为待测组或空白对照组荧光强度值(单位,cps);w为生物组织样品的重量(固态块体组织,单位g)或体积(液态组织,单位ml)。再对t值进行显著性差异分析。当p≥0.05说明待测组与空白对照组不具有显著性差异,判定待测组中不含re-ncap(含量为0);当p<0.05说明待测组与空白对照组具有显著性差异,继续执行步骤(c)和步骤(d)确定待测组中re-ncap的含量。
按照上述方案,步骤(c)具体方法如下:将一定浓度的eu-ncap水悬浮液(0.25μg/ml)分别与空白对照组匀浆、酸溶液按照一定比例(体积比1.52:1)混合均匀,静置一段时间(4h左右)后离心分离得到上清液,将上清液与酸溶液按照一定比例(体积比1:19)混合后得到上清稀释液,将上清稀释液与荧光增强液按照一定比例(体积比1:199)混合后得到测试液,在特定激发波长下测定特定发射波长的荧光强度。空白对照组匀浆的荧光强度记为a1(单位cps),酸溶液的荧光强度a0(单位cps),根据两个荧光强度按照下述公式计算单位质量或者体积组织滞留率s:
其中,s为单位质量或者体积组织滞留率,无量纲;w0为空白对照组生物组织重量或者体积(单位g或ml)。
由单位质量或者体积组织滞留率s按照下述公式计算待测组组织提取率r:
r=(1-s·w1)×100%,
其中,r为组织提取率,无量纲;w1为待测组组织样品的重量或者体积(单位g或ml)。
按照上述方案,步骤(d)中按照下述公式计算待测组生物组织样品中re-ncap的含量m:
其中,m单位nmol/g或者nmol/ml;t1为待测组的单位质量或单位体积组织荧光强度值(单位cps/g或cps/ml);
按照上述方案,所述稀土离子具体为eu3+或tb3+,稀土离子掺杂方式为单独掺杂,或者与其他稀土元素共掺杂。
按照上述方案,re-ncap中re/(re+ca)的摩尔比取值范围为0.1%-18%。
按照上述方案,所述酸溶液具体为硝酸水溶液或者盐酸水溶液,其浓度为0.5-5mol/l。
按照上述方案,所述待测组生物组织样品和对照组生物组织样品选自动物(如鼠、兔、狗等)或人的血液、心脏、肝脏、脾脏、肺脏、肾脏、胰腺、脑、淋巴、骨骼、皮肤、神经、肿瘤等,或者这些动物和人的排泄物(包括尿液、粪便等)。
本发明基于稀土离子可掺杂到ncap结构中、稀土离子自身稳定的荧光特性以及对生物体没有放射性损害等特点,通过酸(如硝酸或盐酸)溶解re-ncap提取出稀土离子,结合稀土离子的荧光强度-浓度标准曲线计算出稀土离子的浓度,最终结合待测组单位质量或者体积组织荧光强度值(t1)、空白对照组的单位质量或者单位体积组织荧光强度的平均值
与现有技术相比,本发明的有益效果主要体现在以下几个方面:1)基于re-ncap自身含有的荧光稀土离子定量检测,可实现re-ncap在各种生物组织及排泄物中的定量检测;2)准确性高,检出限低(0.1nmol/l),检测所需样品量少(10μl左右),检测周期短;3)安全无污染,该方法属于一种安全高效、在常规条件下即可进行的新型检测方法。
附图说明
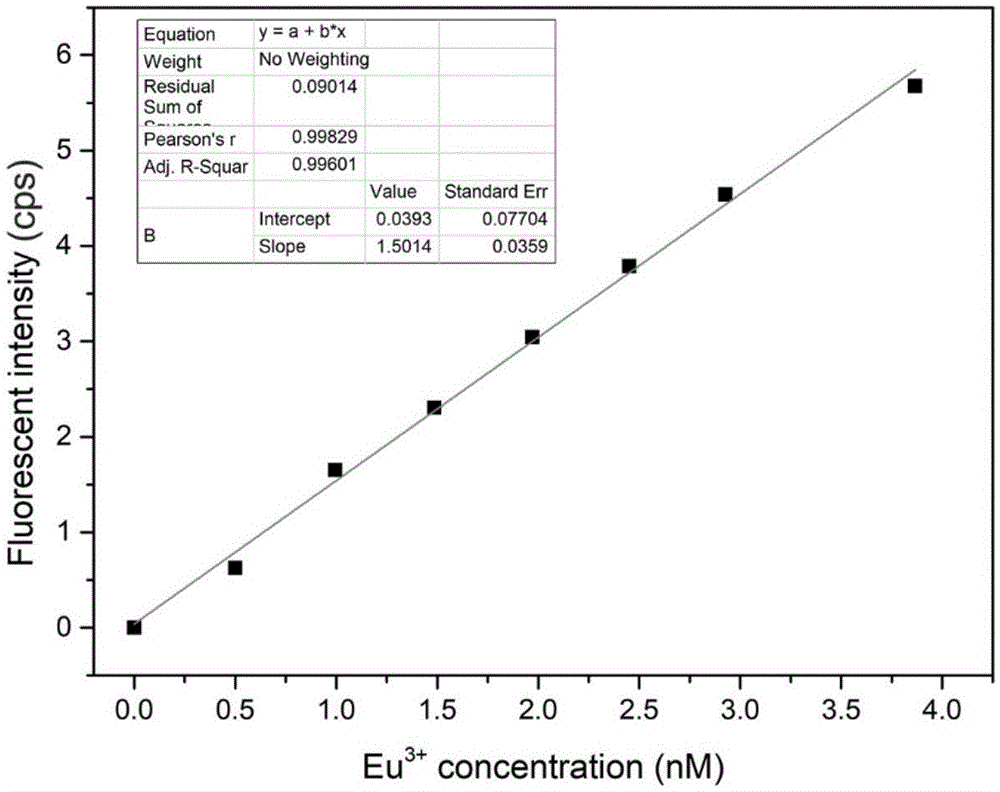
图1为本发明建立的eu离子荧光强度-浓度标准曲线;
图2为实施例1中待测组与空白对照组的单位质量肝脏荧光强度值的检测结果;
图3为实施例2中待测组与空白对照组的单位质量肾脏荧光强度值的检测结果;
图4为实施例3中待测组与空白对照组的单位质量胰腺荧光强度值的检测结果;
图5为实施例4中待测组与空白对照组的单位体积血液荧光强度值的检测结果;
图6为实施例5中待测组与空白对照组的单位重量心脏荧光强度值的检测结果;
图7为实施例6中待测组与空白对照组的单位质量肿瘤的荧光强度值的检测结果。
具体实施方式
为使本领域普通技术人员充分理解本发明的技术方案和有益效果,以下结合具体实施例进行进一步说明。
本发明所使用的铕离子掺杂羟基磷灰石纳米颗粒(eu-ncap,eu/(eu+ca)摩尔比为4%,化学分子式为eu0.4ca9.6(po4)6(oh)2),按照中国专利cn105018086a(稀土掺杂磷酸钙荧光纳米粒子及其制备方法,韩颖超等)、cn107573938a(一种稀土掺杂磷灰石荧光纳米点的制备方法和应用,韩颖超等)公开的方法制备得到。其它稀土离子(如tb)掺杂磷酸钙纳米颗粒可参照该方法制备得到。
本发明涉及的生物组织样品(肝、肾、胰、心脏、血液及肿瘤等)均来自裸鼠,所有裸鼠实验均严格按照国家《实验动物管理条例》及国际相关法律法规的规定执行。实施例1-5均采用体重约20g正常裸鼠进行实验,实施例6采用皮下移植瘤模型(hepg2细胞)的裸鼠(体重约20g)进行实验,肿瘤的大小均为200-300mm3。每个实施例均采用同一批次的6只裸鼠,随机分成2组,分别记为待测组(实验组)和空白对照组。本发明所使用的荧光增强液为the
本发明建立稀土离子荧光强度-浓度标准曲线的方法具体如下:精确称取36.641mg六水合三氯化铕,将其溶于100ml超纯水中,得到浓度为1mmol/l的铕离子储存液。用荧光增强液将铕离子储存液分别稀释成不同浓度(0.50000、0.99502、1.48522、1.97044、2.45098、2.92654、3.86473,单位nmol/l)的铕离子溶液,接着用荧光分光光度计测定不同浓度铕离子溶液在激发波长为340nm条件下618nm处的荧光强度。在直角坐标系中绘制荧光强度y与浓度x的关系曲线,线性拟合得到y=b+nx(r2=0.9960),b为截距,单位cps,其值为0.039;n为斜率,单位cps/(nmol/l),其值为1.501,结果如图1所示。本发明各实施统一使用该标准曲线。
实施例1
通过尾静脉注射将eu-ncap水悬浮液(200μl,5mg/ml)注射到待测组裸鼠体内,立即开始计时。空白对照组裸鼠不进行上述注射处理。8h后将待测组和空白对照组裸鼠的肝脏组织取出,用pbs溶液冲洗干净,每组得到3个肝脏样品(即待测组3个,空白对照组3个)。用纸吸干表面水分后称量每个肝脏样品的重量w(g)。其中待测组w1依次为0.097,0.070,0.074g;空白对照组w0依次为0.054,0.102,0.085g。用2mol/l硝酸将待测组和空白对照组组织样品制成1ml肝脏匀浆。
分别取0.5ml待测组和空白对照组肝脏匀浆,放入离心管中并做好标记,高速离心(5000r/min),取40μl上清液与760μl硝酸(2mol/l)混合制成上清稀释液。取5μl上清稀释液与995μl荧光增强液混合制成混合液,测试混合液在发射波长为618nm处的荧光强度(ex=340nm)。待测组的荧光强度值y1依次为10.068,11.244,10.463cps;空白对照组的荧光强度值y0依次为0.780,0.080,0.653cps。按照公式(i)计算待测组和空白对照组的单位质量肝脏荧光强度值(t):
待测组单位质量肝脏荧光强度值t1依次为103.392,160.071,140.865cps/g;空白对照组单位质量肝脏荧光强度值t0依次为13.722,0.402,7.224cps/g。单因素方差分析可知,待测组t1值与空白对照组t0值相比具有显著性差异(p=0.0017<0.05),结果参见图2。由此可知,需进行后续步骤确定待测组生物组织样品中eu-ncap的含量。
eu-ncap在肝脏中的提取率计算:将相同体积(500ul)的空白对照组匀浆、硝酸(2mol/l)分别放入离心管中,各向其中加入760μleu-ncap悬浮液(0.25μg/ml),混合均匀后放置4h,离心。取40μl上清液与760μl硝酸(2mol/l)混合制成上清稀释液。取5μl上清稀释液与995μl荧光增强液混合制成测试液。测试其在发射波长为618nm处的荧光强度(ex=340nm)。其中空白对照组匀浆的荧光强度a1为2.375cps;硝酸组的荧光强度a0为2.421cps。按照公式(ii)计算单位质量组织滞留率s:
由此计算出的s=0.085,进而按照公式(iii)计算组织提取率r:
r=(1-s·w1)×100%,(1ii)
计算出上述3个待测组的肝脏提取率r依次为97.8%,98.4%,98.3%。
eu-ncap在待测组肝脏中的含量计算:根据已知的荧光强度-浓度标准曲线、待测组的单位质量肝脏荧光强度值(t1)、空白对照组的单位质量肝脏荧光强度值的平均值
本实施例中k为4000/0.4=104,v=1×10-3l,
实施例2
按照实施例1的方法分别得到待测组和对照组裸鼠肾脏,每组3个样品(待测组3个,空白对照组3个)。用天平称量每个肾脏样品的重量w(g),其中待测组w1依次为0.073,0.054,0.065g;空白对照组w0依次为0.020,0.161,0.097g。用2mol/l硝酸将待测组和空白对照组组织样品制成1ml肾脏匀浆。
按照实施例1的方法将待测组和空白对照组肾脏匀浆制成混合液并测试其荧光强度值y,待测组的荧光强度值y1依次为4.555,2.822,2.498cps;空白对照组的荧光强度值y0依次为0.282,0.174,0.095cps。由公式(i)计算出待测组的t1值依次为61.863,51.537,37.831cps/g;空白对照组的t0值依次为1.215,0.839,0.577cps/g。单因素方差分析可知,待测组t1值与空白对照组t0值相比具有显著性差异(p=0.0021<0.05),结果参见图3。由此可知,需进行后续步骤进一步计算待测组中eu-ncap的含量。
按照实施例1的方法测试空白对照组匀浆和硝酸的荧光强度值并根据公式(ii)计算滞留率s,进而根据公式(iii)计算eu-ncap在肾脏中的提取率r。其中a1=2.279cps;a0=2.421cps;w0=0.097g。由此计算出上述3个待测组的肾脏提取率r依次为95.6%,96.7%,96.1%。
eu-ncap在待测组肾脏中的含量计算:按照公式(iv)(本实施例中b=0.039,n=1.501,v=1×10-3l,k=104,
实施例3
按照实施例1的方法分别得到待测组和对照组裸鼠胰腺,每组3个样品(待测组3个,空白对照组3个)。用天平称量每个胰腺样品的重量w(g),其中待测组w1依次为0.027,0.029,0.028g;空白对照组w0依次为0.050,0.033,0.034g。用2mol/l硝酸将待测组和空白对照组组织样品制成1ml胰腺匀浆。
按照实施例1的方法将待测组和空白对照组胰腺匀浆制成混合液并测试其荧光强度值,待测组的荧光强度值y1依次为1.245,1.256,1.320cps;空白对照组的荧光强度值y0依次为0.312,0.491,0.495cps。由公式(ⅰ)可计算出待测组的t1值依次为21.865,28.556,31.615cps/g;空白对照组的t0值依次为8.892,4.148,7.773cps/g。单因素方差分析可知,待测组t1值与空白对照组t0值相比具有显著性差异(p=0.0003<0.05),结果参见图4。由此可知,需进行后续过程进一步确定待测组中eu-ncap的含量。
按照实施例1的方法测试空白对照匀浆和硝酸的荧光强度值并根据荧光强度值计算滞留率s,进而计算eu-ncap在胰腺中的提取率r。其中a1=2.032cps;a0=2.421cps;w0=0.050g。由公式(iii)可计算出上述3个待测组的胰腺提取率r依次为91.32%,90.68%,91.00%。
eu-ncap在待测组胰腺中的含量计算:按照公式(iv)(本实施例中b=0.039,n=1.501,v=1×10-3,k=104,
实施例4
参照实施例1的方法分别采集待测组和对照组裸鼠的血液0.04ml(w),将其与1ml硝酸(2mol/l)混合摇匀制得血液匀浆。每组3个样品(待测组3个,空白对照组3个)。
按照实施例1的方法测试待测组和空白对照组血液匀浆的荧光强度,待测组的荧光强度y1=2.642,2.720,2.810cps;空白对照组的荧光强度y0=0.210,0.241,0.196cps。由公式(ⅰ)可计算出待测组的t1值依次为21.865,28.556,31.615cps/ml;空白对照组的t0值依次为8.892,4.148,7.773cps/ml。单因素方差分析可知,待测组t1值与空白对照组t0值相比具有显著性差异(p=0.000001<0.05),结果参见图5。由此可知,需进行后续过程进一步确定待测组中eu-ncap的含量。
按照实施例1的方法测试空白对照匀浆和硝酸的荧光强度值并根据荧光强度值计算滞留率s,进而计算eu-ncap在血液中的提取率r。其中a1=2.323cps;a0=2.421cps;w0=0.04ml。由公式(iii)可计算出上述3个待测组的血液提取率r都为98.9%。
eu-ncap在待测组血液中的含量计算:按照公式(iv)(本实施例中b=0.039,n=1.501,v=1.04×10-3,k=104,
实施例5
按照实施例1的方法分别得到待测组和对照组裸鼠心脏,每组3个样品(待测组3个,空白对照组3个)。用天平称量每个心脏样品的重量w(g),其中待测组w1依次为0.027,0.024,0.012g;空白对照组w0依次为0.018,0.063,0.020g。用2mol/l硝酸将待测组和空白对照组组织样品制成1ml心脏匀浆。
按照实施例1的方法将待测组和空白对照组心脏匀浆制成混合液并测试其荧光强度值,待测组的荧光强度值y1依次为0.133,0.410,0.120cps;空白对照组的荧光强度值y0依次为0.475,0.117,0.487cps。由公式(ⅰ)可计算出待测组的t1值依次为21.865,28.556,31.615cps/g;空白对照组的t0值依次为8.892,4.148,7.773cps/g。单因素方差分析可知p=0.416>0.05,说明待测组t1值与空白对照组t0值相比不具有显著性差异,结果参见图6。由此判定在心脏组织中的eu-ncap含量为0nmol/g。
实施例6
按照实施例1的方法分别得到待测组和对照组裸鼠肿瘤组织,每组3个样品(待测组3个,空白对照组3个)。用天平称量每个肿瘤样品的重量w(g),其中待测组w1依次为0.037,0.027,0.026g;空白对照组w0依次为0.037,0.115,0.022g。用2mol/l硝酸将待测组和空白对照组组织样品制成1ml肿瘤匀浆。
按照实施例1的方法将待测组和空白对照组肿瘤匀浆制成混合液并测试其荧光强度值,待测组的荧光强度值y1依次为0.848,0.810,0.861cps;空白对照组的荧光强度值y0依次为0.368,0.516,0.210cps。由公式(ⅰ)可计算出待测组的t1值依次为21.865,28.556,31.615cps/g;空白对照组的t0值依次为8.892,4.148,7.773cps/g。单因素方差分析可知,待测组t1值与空白对照组t0值相比具有显著性差异(p=0.0032<0.01),结果参见图7。由此可知,需进行后续过程进一步确定待测组中eu-ncap的含量。
按照实施例1的方法测试空白对照匀浆和硝酸的荧光强度值并根据荧光强度值计算滞留率s,进而计算eu-ncap在肿瘤中的提取率r。其中a1=2.323cps;a0=2.421cps;w0=0.115g。由公式(iii)可计算出上述3个待测组的肿瘤提取率r依次为98.70%,99.05%,99.08%。
eu-ncap在待测组肿瘤中的含量计算:按照公式(iv)(本实施例的b=0.039,n=1.501,v=1×10-3,k=104,
- 还没有人留言评论。精彩留言会获得点赞!