采用一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法与流程
本发明涉及中药成分检测技术领域,具体涉及一种采用一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法。
背景技术:
山苦荬(ixeridiumchinense(thunb.)tzvel.)为菊科小苦荬属多年生草本植物,全草入药,别名中华苦荬菜、山苦菜、小苦菜等,分布在北部、南部及东部省区,既可药用又可食用,具有清热解毒、消炎凉血、止痛消肿、抗肿瘤等功效,用于治疗无名肿痛、腹腔脓肿、痢疾、阑尾炎、肺炎、关节炎等症。
刘畅等对山苦菜的化学成分进行深入系统的研究发现山苦菜中含有黄酮类成分,其中黄酮类成分具有很强的药理活性;主要含有:木犀草素,木犀草素-7-o-β-d-葡萄糖苷,芹菜素-7-o-β-d-葡萄糖苷,黑麦草素,木犀草素-7-o-β-d-葡萄糖苷乙酸酯,芹菜素,还含有咖啡酸等(刘畅,严铭铭,邵帅,等.山苦菜化学成分及药理作用的研究进展.中国实验方剂学杂志,2015)。周桃等为了更好地利用山苦荬资源,建立了测定山苦荬中木犀草苷含量的hplc法,并测定了6个不同产地山苦荬中木犀草苷的含量,得到6个不同产地山苦荬中木犀草苷的含量为1.2812.288mg·g-1,平均含量为1.780mg·g-1(周桃,隋琳,那生桑,等.hplc法测定不同产地山苦荬中木犀草苷的含量[j].今日药学,2018)。周宏雷等对中华苦荬菜别名(山苦荬)的化学成分进行研究,从醇提物中分离得到5种化合物(周宏雷,袁久荣.中华苦荬菜化学成分的研究[j].中草药,1996)。
药理研究方面,太文杰等得出山苦荬具有多种药理活性,包括抗炎保肝作用,抗病毒、抗白血病作用,治疗糖尿病作用等;由化学成分研究可知,山苦荬中含有多种黄酮类化合物,具有抗肿瘤、抗氧化等,并在最佳提取工艺条件下,提取获得山苦荬总黄酮,并采用可见分光光度法测定山苦荬总黄酮的含量,结果表明芦丁浓度范围内呈良好的线性(太文杰,陈生科,甘秀海.山苦荬中总黄酮的提取及抗氧化活性研究[j].贵州师范学院学报,2016)。周黎等初次报道了中华苦荬菜别名(山苦荬)提取物chinensiolidea在体外可有效的抑制肺腺癌a549细胞、肝癌ble-7402细胞及lovo细胞的生长,具备较强的抗肿瘤活性(周黎,赵颖,王玉,等.中华苦荬菜提取物chinensiolidea的体外抗肿瘤活性[j].中国医学创新,2015)。王璐璐等在研究中发现,中华苦荬菜别名(山苦荬)具有良好的抗病毒及降低血脂和胆固醇等作用(王璐璐,杨莉,李岳桦,等.中华苦荬菜总黄酮和总三萜提取工艺优化研究[j].广东化工,2011)。程梁华等在研究中华苦荬菜别名(山苦荬)中发现发现中华苦荬菜提取物有显著的降血糖作用;在本次实验的过程中发现含有绿原酸成分,并对该成分进行研究和探索;说明山苦荬含有大量的黄酮类成分,并起到很大的药理治疗作用(程梁华.中华苦荬菜总黄酮提取物的制备和治疗2型糖尿病的药效及机制研究[d].黑龙江中医药大学,2016)。
上述可见,山苦荬是我国中药中重要且常用的一味,药用价值很高,而其质量的好坏与人们的健康利益密切相关。影响中药材质量因素很多,导致市场上中药材质量控制困难,目前仅凭单一成分评价中药材质量过于片面,不能很好反应中药材质量优良。由于植物化学成分的复杂性,多指标含量测定已经成为植物来源药物质量控制的共识。以传统测定方法同时测定中药材中几种成分,不但面临对照品难得,检验周期长,还耗能、耗时。一测多评法通过中药有效成分间存在的内在函数关系和比例关系法,测定一个成分(对照品易得、廉价、有效者)实现多个成分(对照品难以得到或难供应者)同步测定;不仅解决了中药多成分定量和多指标质量控制中存在的对照品缺乏问题,同时实现多指标同步质量控制反应中药质量。目前关于山苦荬的质量分析的相关研究较少,经查阅文献,当前国内外尚无一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的报道。
技术实现要素:
本发明的目的是针对现有技术的不足提供一种采用一测多评法测定山苦荬(ixeridiumchinense(thunb.)tzvel.)中绿原酸、咖啡酸和木犀草苷含量的方法,该方法简单、准确,操作方便,实用性高,结果准确,采用该方法能实现山苦荬中绿原酸、咖啡酸和木犀草苷成分含量的同步测定,从而有效评价和控制山苦荬(ixeridiumchinense(thunb.)tzvel.)药材的质量,为保证临床用药安全和有效,及其药材资源的开发提供科学依据。
为实现上述目的,本发明采用如下技术方案:
采用一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法,包括如下步骤:
(1)对照品溶液的制备:精密称量绿原酸对照品11.59mg,咖啡酸对照品6.72mg、木犀草苷对照品13.60mg,分别加入25ml容量瓶中,加适量甲醇稀释至刻度,超声使其溶解,摇匀,精密量取上述绿原酸溶液1.5ml、咖啡酸溶液1.5ml和木犀草苷溶液6.0ml,置同一10ml容量瓶中并用甲醇定容至刻度,摇匀,即得混合对照品溶液,混合对照品溶液每1ml溶液中含绿原酸66.9μg,咖啡酸40.35μg,木犀草苷306.78μg;
(2)供试品溶液的制备:取山苦荬药材粉末2g,置于100ml锥形瓶中,加入体积浓度为70%的甲醇30ml,称定重量,超声提取60min,温度30℃,频率70khz,放冷,用体积浓度为70%的甲醇补足减失的重量,过滤,取续滤液过微孔滤膜0.45µm,即得;
(3)色谱条件与系统适用性试验:色谱柱:capcellpaktypemgⅱsize-c18(4.6×250mm,5μm);体积流量为1.0ml·min-1;检测波长为330nm;柱温为25℃;进样量为5µl;流动相采用甲醇-0.2%磷酸水溶液进行梯度洗脱,洗脱程序见表1,理论塔板数按绿原酸峰计算应不低于5000;
(4)测定:分别精密吸取对照品溶液与供试品溶液各5µl,注入液相色谱仪,在上述系统适应性的色谱条件下测定,采用内标法计算,供试品溶液中绿原酸、咖啡酸和木犀草苷色谱峰与对照品色谱峰保留时间一致,并与其它共存成分的分离度大于1.5,主峰与杂质峰达到基线分离;供试品溶液的特征图谱中呈现绿原酸、咖啡酸和木犀草苷的峰值,以绿原酸为内参物,计算绿原酸与咖啡酸和木犀草苷的相对校正因子,通过相对校正因子计算山苦荬中绿原酸、咖啡酸和木犀草苷含量。
上述的采用一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法,所述的相对校正因子的计算方法为:分别吸取步骤(1)制备得到的混合对照品溶液0.4、0.6、0.8、1.0、1.2、1.6ml,分别置2.0ml量瓶中,加体积浓度为70%的甲醇定容至刻度,摇匀,制成不同浓度的系列混合对照品溶液,按色谱条件测定,分别进样5μl,以绿原酸为内标,测定山苦荬中咖啡酸、木犀草苷的峰面积,按照相对校正因子计算公式fac=fa/fc=(aa/ca)/(ac/cc),计算常咖啡酸、木犀草苷的相对校正因子fac,式中用对照品来计算,aa为内参物绿原酸的峰面积,ca为内参物绿原酸的浓度,ac为某待测成分峰面积;cc为某待测成分浓度;相对校正因子fac为各实验所得数据的平均值。
本发明的有益效果为:
1、本发明采用一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法,符合方法学验证的要求,在对照品短缺的情况下,本发明的方法可以应用于山苦荬的质量评价,同时测定山苦荬中绿原酸、咖啡酸和木犀草苷的含量,不仅简便、快捷,而且重复性好,可以节约资源的损耗,减少溶剂消耗和分析时间,更有利于环境保护,为建立金花草药材的质量评价提供依据。
2、本发明提供的一种采用一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法,该方法简单、测定准确,操作方便,实用性高,结果准确,方法重复性,耐用性好,采用该方法能实现山苦荬中绿原酸、咖啡酸和木犀草苷成分含量的同步测定,从而可以更好、更全面有效的评价和控制山苦荬药材的质量,能够用于对山苦荬药材的质量控制,为保证临床用药安全和有效,及其药材资源的开发提供科学依据;同时规避由多成含量测定带来的成本和操作问题。
附图说明
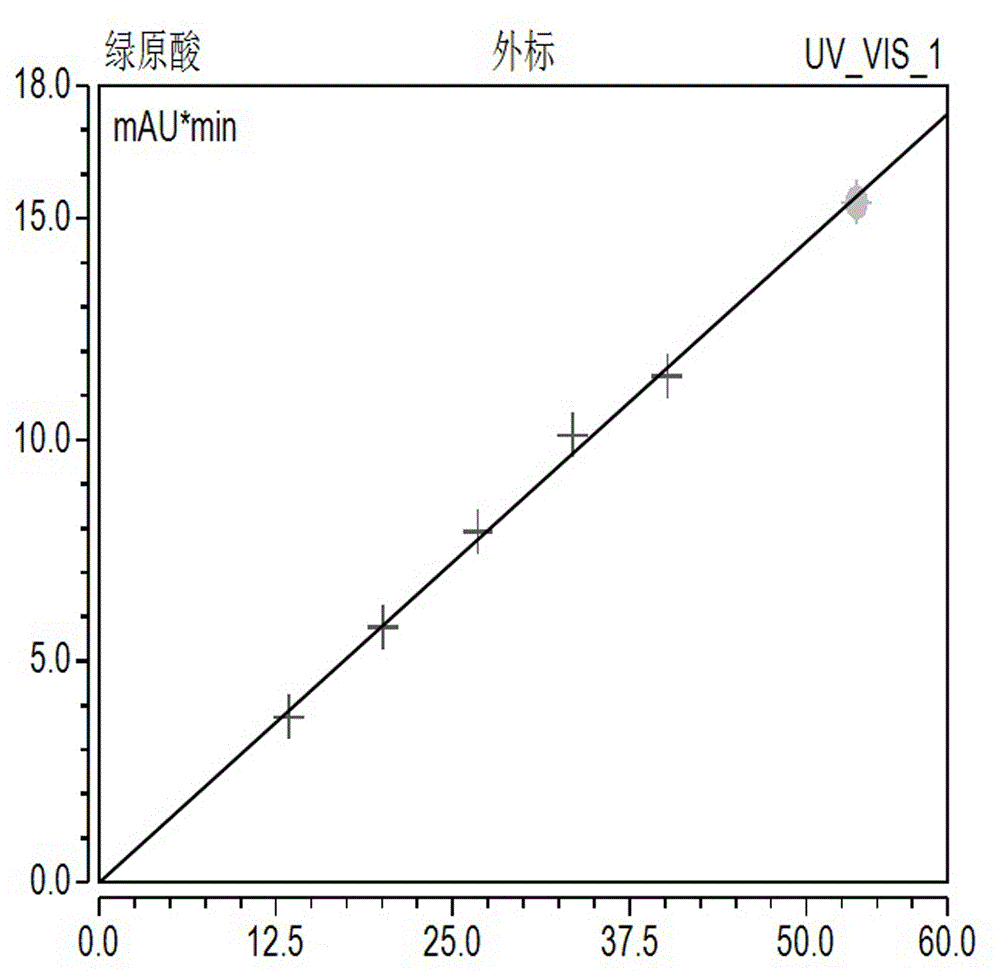
图1为绿原酸标准曲线图;
图2为咖啡酸标准曲线图;
图3为木犀草苷标准曲线图;
图4为混合对照品色谱图,图中,1、绿原酸,2、咖啡酸,3、木犀草苷;
图5为山苦荬样品色谱图,1、绿原酸,2、咖啡酸,3、木犀草苷。
具体实施方式
实施例1
采用上述一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法对10批山苦荬药材进行检测,具体测定情况如下:
1实验材料、仪器与试药
1.1实验材料
山苦荬中药材采收地区及日期见表2,经广西中医药大学韦松基教授鉴定为菊科小苦荬属植物山苦荬(ixeridiumchinense(thunb.)tzvel.)的全草。
1.2仪器
u3000高效液相色谱仪(美国thermofisher公司);1260高效液相色谱议(美国agilent公司);色谱柱capcellpaktypeⅱsizec18(4.6mmi.d.×250mm,5µm),agilenteclipseplusc18(4.6×250mm,5µm),watersatlantist3(4.6×250mm,5µm);xs-205du电子分析天平(瑞士mettlertoledo公司);kq-500gdv超声波清洗器(昆山市超声仪器有限公司);501超级恒温水浴锅(金坛市医疗仪器厂);synergy超纯水机(美国millipore公司)。
1.3试药
绿原酸(110753-201415,含量以96.2%计)、咖啡酸(110885-200102)、木樨草苷(111720-201307含量以94.0%计)均购自中国食品药品检定研究院。乙腈、甲醇(色谱纯,美国fisher公司),磷酸(批号:20150327,天津市光复精细化工研究所,优级纯),效液相色谱议用水为超纯水,其他所用试剂均为分析纯。
2实验方法与结果
2.1采用一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法,包括如下步骤:
(1)对照品溶液的制备:精密称量绿原酸对照品11.59mg,咖啡酸对照品6.72mg、木犀草苷对照品13.60mg,分别加入25ml容量瓶中,加适量甲醇稀释至刻度,超声使其溶解,摇匀,精密量取上述绿原酸溶液1.5ml、咖啡酸溶液1.5ml和木犀草苷溶液6.0ml,置同一10ml容量瓶中并用甲醇定容至刻度,摇匀,即得混合对照品溶液,混合对照品溶液每1ml溶液中含绿原酸66.9μg,咖啡酸40.35μg,木犀草苷306.78μg;
(2)供试品溶液的制备:取山苦荬药材粉末2g,置于100ml锥形瓶中,加入体积浓度为70%的甲醇30ml,称定重量,超声提取60min,温度30℃,频率70khz,放冷,用体积浓度为70%的甲醇补足减失的重量,过滤,取续滤液过微孔滤膜0.45µm,即得;
(3)色谱条件与系统适用性试验:色谱柱:capcellpaktypemgⅱsize-c18(4.6×250mm,5μm);体积流量为1.0ml·min-1;检测波长为330nm;柱温为25℃;进样量为5µl;流动相采用甲醇-0.2%磷酸水溶液进行梯度洗脱,洗脱程序见表1,理论塔板数按绿原酸峰计算应不低于5000;
(4)测定:分别精密吸取对照品溶液与供试品溶液各5µl,注入液相色谱仪,在上述系统适应性的色谱条件下测定,采用内标法计算,供试品溶液中绿原酸、咖啡酸和木犀草苷色谱峰与对照品色谱峰保留时间一致,并与其它共存成分的分离度大于1.5,主峰与杂质峰达到基线分离;供试品溶液的特征图谱中呈现绿原酸、咖啡酸和木犀草苷的峰值,以绿原酸为内参物,计算绿原酸与咖啡酸和木犀草苷的相对校正因子,通过相对校正因子计算山苦荬中绿原酸、咖啡酸和木犀草苷含量。
上述的采用一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法,所述的相对校正因子的计算方法为:分别吸取步骤(1)制备得到的混合对照品溶液0.4、0.6、0.8、1.0、1.2、1.6ml,分别置2.0ml量瓶中,加体积浓度为70%的甲醇定容至刻度,摇匀,制成不同浓度的系列混合对照品溶液,按色谱条件测定,分别进样5μl,以绿原酸为内标,测定山苦荬中咖啡酸、木犀草苷的峰面积,按照相对校正因子计算公式fac=fa/fc=(aa/ca)/(ac/cc),计算常咖啡酸、木犀草苷的相对校正因子fac,式中用对照品来计算,aa为内参物绿原酸的峰面积,ca为内参物绿原酸的浓度,ac为某待测成分峰面积;cc为某待测成分浓度;相对校正因子fac为各实验所得数据的平均值。
2.2本发明一测多评法测定山苦荬中绿原酸、咖啡酸和木犀草苷含量的方法的方法学考察如下:
2.2.1线性范围的考察结果
分别吸取本发明步骤(1)制备得到的混合对照品溶液0.4、0.6、0.8、1.0、1.2、1.6ml,分别置2.0ml量瓶中,加70%甲醇定容至刻度,摇匀,制成不同浓度的系列混合对照品溶液,按本发明所限定的色谱条件测定,分别进样5μl,以峰面积为纵坐标,浓度为横坐标经线性回归范围考察见表3,结果见图1、2、3。由结果可得:在本次实验中线性范围考察良好。
2.2.2精密度试验
取s4号山苦荬粉末2g,按本发明供试品溶液的制备方法制备山苦荬供试品溶液,按照色谱条件连续进样六次,记录样品图谱和计算山苦荬峰面积。结果见表4:绿原酸、咖啡酸和木犀草苷平均峰面积值分别为为15.617、5.467和31.040,rsd分别为2.0%、2.1%和2.1%,结果可得仪器的精密度良好。
2.2.3重复性试验
取s4号山苦荬粉末6份,每份2g,按本发明供试品溶液的制备方法制备供试品,按照色谱条件来进行测定,观察图谱峰和记录山苦荬样品数据,计算山苦荬中绿原酸、咖啡酸、木犀草苷的含量。结果见表5:绿原酸平均含量为1.167mg/g,rsd为1.7%,咖啡酸平均含量为0.222mg/g,rsd为2.3%,木犀草苷平均含量为2.510mg/g,rsd为2.3%。表明该方法的重复性良好。
2.2.4稳定性试验
取重复性试验中一供试品溶液,按照色谱条件,在1,2,4,6,8,12,24h进行测定,记录山苦荬样品图谱和峰面积并计算山苦荬测得含量数据,考察供试品溶液的稳定性。结果见表6:绿原酸平均含量1.146mg/g,rsd为5.1%,咖啡酸平均含量为0.213mg/g,rsd为4.8%,木犀草苷平均含量为2.416mg/g,rsd为5.1%。得出在24h内的供试品溶液稳定性良好。
2.2.5加样回收试验
精密称定已测定含量(每1g中含绿原酸1.167mg、咖啡酸0.222mg、木犀草苷2.510mg)的山苦荬样品6份,每份1g;另精密称取绿原酸对照品6.86mg,咖啡酸对照品1.34mg,木犀草苷对照品16.24mg,置200ml量瓶中,加适量70%甲醇超声使溶解,并稀释至刻度,摇匀,即得混合对照品储备液(每1ml中含绿原酸33.0μg、咖啡酸6.7μg、木犀草苷76.3μg)。分别精密加入对照品贮备液30ml,按“2.5”项下制备山苦荬加样回收样品溶液,按照色谱条件测定,并记录绿原酸、咖啡酸、木犀草苷的峰面积并计算回收率,由结果可得rsd<3。结果见表7、8、9。
2.3山苦荬中待测指标相对校正因子的计算
在本发明限定的色谱条件下,精密吸取“2.2.1线性范围的考察结果”项下的混合对照品溶液,以绿原酸为内标,测定山苦荬中咖啡酸、木犀草苷的峰面积,按照相对校正因子计算公式计算常咖啡酸、木犀草苷的相对校正因子,结果见表10,相对校正因子取平均值。按下式计算:fac=fa/fc=(aa/ca)/(ac/cc),式中用对照品来计算,aa和为内参物(绿原酸)峰面积,ca为内参物(绿原酸)浓度,ac为某待测成分峰面积;cc为某待测成分浓度。
2.4系统耐用性的考察
在本发明限定的色谱条件下测定,精密吸取“2.2.1线性范围的考察结果”项下混合对照品溶液,为考察耐用性计算对照品中待测成分含量的相对校正因子、相对保留值,并考察色谱法的定位,可以更好的评测所得到的结果,以验证一测多评法在今后的应用。
2.4.1不同进样量考察
在“2.4”项下,考察4,5,6µl不同进样量的影响,结果见表11。不同体积流量对其影响不显著,其耐用性良好。
2.4.2不同仪器、色谱柱考察
在“2.4”项条件下,选择thermoultimate3000、agilrnt1260,以及ypemgⅱsize-c18、agilenteclipseplus-c18、atlantist3-c18色谱柱(4.6×250mm5µm),考察相对校正因子的影响,结果见表12。可知不同仪器、色谱柱对其影响程度不明显,耐用性良好。
2.4.3不同体积流量考察
在“2.4”项下,考察0.9、1.0、1.1ml/min体积流量对相对校正因子和相对保留值的影响,结果见表13。不同体积流量对其影响程度不明显,耐用性良好。
2.4.4不同柱温考察
在“2.4”项下,考察20、25、30、℃柱温对相对校正因子和相对保留值的影响,结果见表14。不同柱温对其影响程度不明显,耐用性良好。
2.4.5不同波长考察
在“2.4”项下,考察220、254、330nm检测波长对相对校正因子和相对保留值的影响,结果见表15。其检测波长对其影响程度不明显,耐用性良好。
2.5一测多评法待测色谱峰的定位
在“2.4”项下,考察“2.4.2”项下在不同仪器、色谱柱中的相对保留值的影响,结果见表16。由表可知,相对保留值波动较小,咖啡酸和木犀草苷的rsd分别为1.4%和6.8%,因此选择不同仪器、色谱柱相对保留值作为色谱峰定位指标。
2.6一测多评法与外标法含量测定结果比较
经方法学优化和考察后,采用以上建立的一测多评方法,对10批山苦荬提取物中的咖啡酸、木犀草苷成分的含量进行了测定,按“2.2.1”项下对照品溶液,按照本发明的测定方法进样测定,分别测定咖啡酸、木犀草苷的含量(图4、图5)。采用外标法和相对校正因子进行比较,结果见表17。
为验证一测多评法“qams”方法的准确性,对“qams”法的测定结果与外标法测定结果进行了比较。以准确度(accuracy)来评价所建立的一测多评方法的可行性,准确度由以下公式计算:准确度(%)=cs/ce×100%,其中,cs表示一测多评法计算所得的含量,ce表示通过外标法计算所得的含量。
实验结果:在本次实验采用一测多评法测定10批产地山苦荬中绿原酸、咖啡酸和木犀草苷的含量,在结果显示以绿原酸含量较高的产地为s10、s6、s4,咖啡酸含量较高的产地为s10、s5、s6、s4、s2,木犀草苷含量较高的产地为s10、s5、s4、s6,从中可以得出s10、s6、s4产地的山苦荬绿原酸、咖啡酸和木犀草苷的含量较高。
3讨论
3.1流动相的选择
本实验为考察山苦荬提取物的含量,从而确定所含的各成分,经查阅文献可得,选择0.1%磷酸:乙腈;0.1%磷酸:甲醇;0.2%磷酸:乙腈;0.2%磷酸:甲醇;水:甲醇等各种流动性进行考察;实验过程中发现因水的极性较大,出峰成分较少且峰与峰之间堆在一起,所以考虑加酸;用0.1%磷酸时达不到分离效果,故选用0.2%磷酸;在选用乙腈和甲醇时,乙腈在分离时锋形较窄且分离效果较差,选择0.2%磷酸:甲醇采用梯度洗脱时分离效果较好。
3.2供试品制备
通过考察提取条件来确定供试品的制备,在不同提取方法、不同溶剂浓度甲醇、不同料液比和不同提取时间,提取得到的溶液直接过微孔滤膜,过滤后直接进样,实验发现用70%甲醇30ml超声60min后所得到的峰基线平稳,色谱图中检测成分较多且峰形较好。在考察回流法和超声法时发现,两者提取的含量区别不大,查阅文献发现,较多喜欢用超声提取法。
3.3检测波长的选择
本实验在考察样品时确定流动性,色谱条件,采用对照品来确定山苦荬中含有绿原酸、咖啡酸和木犀草苷,并对确定的对照品和药材粉末以确定的色谱条件进行全波长扫描时发现在330nm都有共有峰出现,且保留时间相同,色谱峰形也较好,所以用330nm作为检测波长。
3.4内标筛选
在本次实验中山苦荬由于含有较多黄酮类成分多,在用外标法计算山苦荬中3种含量时发现,绿原酸的含量与3种成分的含量较稳定,在限有对照品的条件下所测得三个含量中绿原酸对照品较易购买,获得性高,以绿原酸为内参物,建立其咖啡酸和木犀草苷的相对校正因子。且实验发现,在不同仪器和色谱柱上绿原酸出峰时间适中且保留时间差波动较小,rsd均在5%以内;由于咖啡酸中含量较低,较不稳定,不宜进行系统性分析;而木犀草苷的保留时间大于5%,故不宜进行各组分之间的定位。所以选择绿原酸为内标,用“qams”法中的相对校正因子算咖啡酸和木犀草苷的含量。
综上所述,本实验建立的含量测定方法操作方便、结果准确,可用于山苦荬中绿原酸、咖啡酸和木犀草苷成分含量的同步测定
3.5“qams”法耐用性的考察
本实验采用“qams”法选择考察ultimate3000和agilent1260,并采用不同的色谱条件,对此方法的耐用性进行考察,所测得各成分对相对校正因子和相对保留值的影响,验证此方法在山苦荬质量控制上的耐用性。结果表明:可在对照品短缺的情况下,采用“一测多评法”中的重复性均较理想,并可以节约资源的损耗。
3.6色谱峰定位
本实验是在只使用单一对照品时,可以采用一测多评法正确定位山苦荬中所测得咖啡酸、木樨草苷2种成分色谱峰的问题,选择各成分之间的保留时间差、相对保留值、分离度作为定位标准,并用不同仪器、色谱柱对二者进行考察。结果表明,相对保留值能更准确地定位色谱峰。
3.7外标法与一测多评法的评价
通过用10批产地山苦荬药材所测得的含量进行分析,用外标法对一测多评法的结果进行比较,得到的结果大致一样,结果表明,一评多测方法简便、准确度高,具有较高的可行性。
4结论
利用外标法和一测多评法进行比较10批山苦荬药材,在结果中的得出,各个产地之前存在的含量有显著差异,不同的地方有些含量较高,有些含量较低,说明了道地药材的重要性;采用该方法简单、准确,操作方便,所以能在对照品短缺的情况下用于含量测定,同时可减少溶剂消耗和分析时间,更有利于环境保护。
- 还没有人留言评论。精彩留言会获得点赞!