一种检测富马酸福莫特罗或其相关制剂中杂质的方法与流程
本发明涉及药物检测
技术领域:
,具体涉及一种检测富马酸福莫特罗或其相关制剂中杂质的方法。
背景技术:
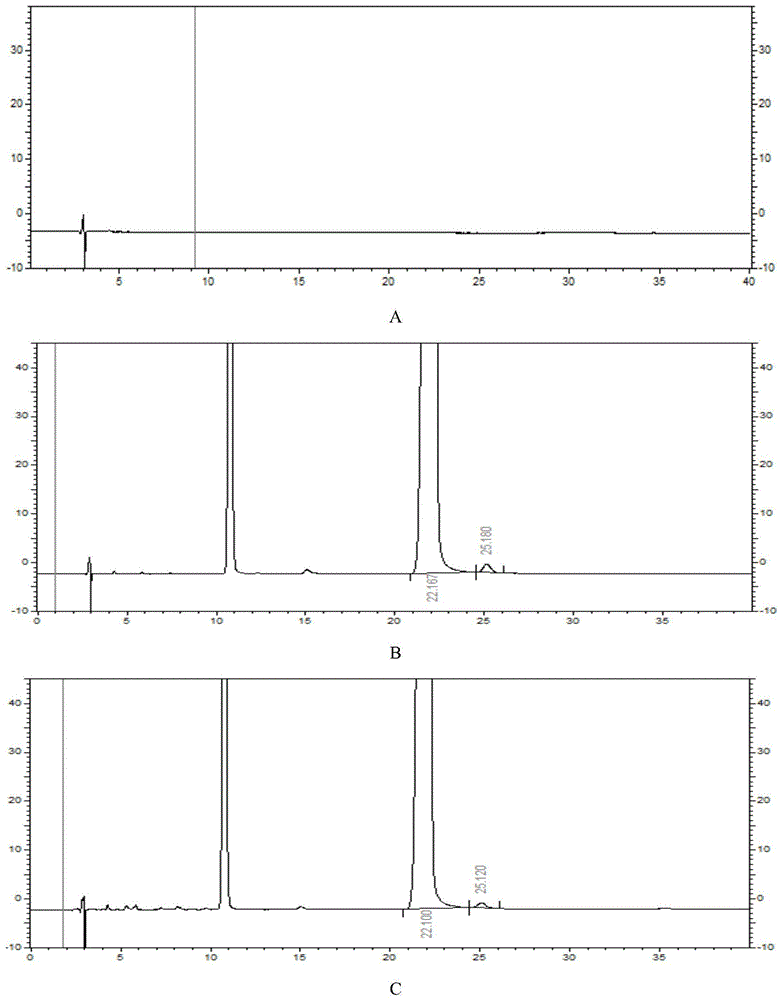
:富马酸福莫特罗(formoterolfumarate),是由日本山之内制药株式会社开发的β2-肾上腺素受体激动剂,用于治疗支气管哮喘,急性支气管炎或喘息性支气管炎等气道阻塞性肺病,化学名:(±)-n-[2-羟基-5-[(1rs)-羟基-2[[(1rs)-2-(4-甲氧基苄基)-1-甲基乙基]氨基]乙基]苯基]甲酰胺富马酸盐二水合物,是福莫特罗(formoterol)的可药用盐,结构如式(1)所示:目前,中国哮喘防治指南、美国胸科协会(ats)以及欧洲呼吸协会(ers)已经把富马酸福莫特罗列为作为控制哮喘的支气管舒张剂的首选长效β2受体激动剂。已上市的富马酸福莫特罗剂型主要为片剂和吸入剂两种,例如:由阿斯利康公司(astrazeneca)生产的富马酸福莫特罗粉雾剂,2004年获批进口中国,商品名“奥克斯都保”,已经成为常用有效的哮喘缓解控制药物。由于福莫特罗分子有两个手性中心,共4种光学构型,其中,作为富马酸福莫特罗药用活性成分的为r,r与s,s构型的一对外消旋体;r,s与s,r构型的两个光学异构体为富马酸福莫特罗的非对映异构体杂质i,国内外药典对富马酸福莫特罗非对映异构体杂质i的限度有很高的要求,例如:欧洲药典ep9.7、中国药典2015版、美国药典usp41均要求富马酸福莫特罗杂质i最大为0.3%。因此,富马酸福莫特罗中非对映异构体杂质的检测是富马酸福莫特罗及其相关制剂的质量研究中关键性项目之一。在现有技术中,由于高效液相色谱(highperformanceliquidchromatography,hplc)法具有简便、灵敏、快速、准确和重现性好等特点,已将其广泛的用于富马酸福莫特罗的含量测定和质量控制,国内外药典均收载了富马酸福莫特罗非对映异构体检测方法,例如:中国药典2015年版收载的富马酸福莫特罗原料药中非对映异构体检测方法,其采用高效液相色谱法、十八烷基键合聚乙烯醇为固定相,以磷酸盐缓冲液(用5mol/l的氢氧化钾或者磷酸调节ph至12.0±0.1)和乙腈体积比=88:12为流动相,柱温30℃,检测波长225nm,且美国药典41版和欧洲药典9.7也均采用该方法对富马酸福莫特罗非对映异构体进行检测。然而,上述方法在实际运用中存在以下不足:1)以十八烷基键合聚乙烯醇为固定相的色谱柱在该方法中耐受性差,使用寿命短;2)由于十八烷基键合聚乙烯醇为固定相的色谱柱在该方法中耐受性差,导致在色谱柱长时间使用后,检测结果的重现性差;3)目前,以碳十八聚乙烯醇为固定相的色谱柱,其生产厂家只有日本昭和电工株式会社,价格昂贵,可替代产品稀缺,企业使用成本高,制约行业的发展。因此,在现有技术公开的富马酸福莫特罗非对映异构体检测方法中,存在色谱柱使用寿命短,检测结果重现性差,方法应用成本高的缺陷。另外,本领域缺少可检测富马酸福莫特罗相关制剂中非对映异构体的方法。技术实现要素:本发明的目的是提供一种检测富马酸福莫特罗或其相关制剂中非对映异构体杂质的方法。本发明采用高效液相色谱法,采用离子对试剂缓冲液-有机相进行等度洗脱,色谱柱耐受性好、使用寿命长,检测结果准确、重现性好,且可选色谱柱类型多,降低了方法应用成本。采用本发明的方法得到的测定结果准确,并能同时适用于检测富马酸福莫特罗原料药及其相关制剂中非对映异构体杂质的含量。为了实现上述发明目的,本发明采用的技术方案如下:一种检测富马酸福莫特罗或其相关制剂中杂质的方法,该方法包括采用高效液相色谱法检测富马酸福莫特罗或其相关制剂中的非对映异构体杂质(杂质i)的步骤,其中,所述高效液相色谱中使用的洗脱剂为由a液和b液组成的体系,所述a液为离子对试剂缓冲液,所述b液为乙腈,洗脱方式为等度洗脱;优选地,所述a液为四丁基氢氧化铵水溶液;优选地,所述a液的浓度为0.3%~0.7%(v/v);更优选地为0.6%(v/v);优选地,采用磷酸来调节所述a液的ph值;更优选地,将所述a液的ph值调节为7.0~10.0,更优选地调节为8.5;优选地,所述a液与所述b液的体积比为(85:15)~(75:25),更进一步优选为80:20;优选地,所述方法的洗脱流速为0.8ml/min~1.2ml/min,进一步优选为1.0ml/min;优选地,所述方法的检测柱温为25~50℃;进一步优选为35℃;优选地,所述方法采用的固定相为硅烷键合相;进一步优选为碳八烷基硅烷键合相或碳十八烷基硅烷键合相;更进一步优选为碳十八烷基硅烷键合相;优选地,所述方法中采用的色谱柱选自agilentzorbaxsb-c8和watersxbridgec18色谱柱;更优选地为:watersxbridgec18色谱柱;优选地,所述方法的检测波长为210~260nm;进一步优选为225nm;优选地,所述方法的进样量为20~50μl;进一步优选为50μl。作为一个优选的具体实施方式,本发明提供一种高效液相色谱法检测富马酸福莫特罗或其相关制剂中非对映异构体杂质的方法,该方法包括如下色谱条件:高效液相色谱仪:安捷伦:aglient1260(dad);固定相:碳十八烷基硅烷键合相,粒径5μm;流动相:a液为用磷酸调节ph为8.5的四丁基氢氧化铵水溶液(0.6%,v/v),b液为乙腈;a液:b液=80:20(v/v);流速:1.0ml/min;柱温:35℃;紫外检测器,检测波长220nm;进样量:50μl;优选地,所述方法还包括空白溶液(稀释液)的配制:分别量取乙腈120ml和水880ml,摇匀,即得;优选地,所述方法还包括系统适用性溶液的配制:称取富马酸福莫特罗杂质i鉴别用对照品(edqm,欧洲官方对照品)约2mg,置20ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,即得;优选地,所述方法还包括原料药供试品溶液的配制:精密称取富马酸福莫特罗原料药7mg,置于100ml容量瓶中,加稀释液稀释溶解并稀释至刻度,摇匀,即得;优选地,所述方法还包括制剂供试品溶液的配制(富马酸福莫特罗气雾剂,120揿/瓶,每揿12μg):取富马酸福莫特罗气雾剂1瓶,在室温下倒置(阀杆朝下)1小时,采用尖头锥在样品罐底部扎一小孔,逸去抛射剂后,用割管器除去样品罐底部,用稀释液将内容物溶解,转移至20ml量瓶中,加稀释液至刻度,摇匀,即得;优选地,所述方法还包括如下测定方法:分别吸取空白溶液、系统适用性溶液与供试品溶液各50μl,注入高效液相色谱仪,记录色谱图,测量供试品溶液色谱图上各吸收峰的峰面积,用峰面积归一化法计算杂质i的含量。以下对富马酸福莫特罗非对映异构体杂质进行说明:福莫特罗分子有两个手性中心,共4种光学构型,需要严格限定含量的杂质有:具有如(2)所示结构的r,s构型杂质,具有如(3)所示结构的s,r构型杂质,统称为杂质i。本发明所述测定方法相对于现有技术的优点在于:1)本发明的方法采用离子对试剂缓冲液-有机相进行等度洗脱,基线平稳,主峰峰形良好,主峰与杂质峰之间能够实现良好的分离;2)本发明所述方法具有优良的柱与柱之间的重现性;3)本发明所述方法适用范围宽,流动相a液的浓度在0.3%~0.7%(v/v)、ph7.0~10.0、流动相比例在(85:15)~(75:25)的范围内都可以用于测定。4)本发明所述方法以硅烷键合相为固定相,色谱柱的耐受性好,使用寿命长;5)可适用的色谱柱种类多,价格低廉,大大降低了企业的使用成本。6)本发明所述方法同时适用于检测富马酸福莫特罗原料药及其相关制剂中非对映异构体杂质的含量,弥补了现有技术中检测富马酸福莫特罗制剂中非对映异构体杂质含量的空白。附图说明图1是实施例1的hplc图谱,其中图1a为空白溶液图谱,图1b为系统适用性溶液图谱,图1c为富马酸福莫特罗吸入气雾剂供试品溶液图谱。图2是实施例2的hplc图谱,其中,图2a为杂质a定位溶液,图2b为杂质i定位溶液,图2c为富马酸福莫特罗吸入气雾剂供试品溶液图谱。图3是实施例3的hplc图谱,图3a为空白溶液图谱,图3b为系统适用性溶液图谱,图3c为富马酸福莫特罗原料药供试品溶液图谱。图4是实施例4的hplc图谱,其中,图4a为0小时系统适用性溶液,图4b为40小时系统适用性溶液。图5是实施例5的hplc图谱,其中,图5a为25℃系统适用性溶液图谱,图5b为35℃系统适用性溶液图谱,图5c为40℃系统适用性溶液图谱,图5d为45℃系统适用性溶液图谱,图5e为50℃系统适用性溶液图谱。图6是实施例6的hplc图谱,其中,图6a为0.2%(v/v)系统适用性溶液图谱,图6b为0.3%(v/v)系统适用性溶液图谱,图6c为0.6%(v/v)系统适用性溶液图谱,图6d为0.7%(v/v)系统适用性溶液图谱。图7是实施例7的hplc图谱,其中,图7a为ph6.0系统适用性溶液图谱,图7b为ph7.0系统适用性溶液图谱,图7c为ph8.5系统适用性溶液图谱,图7d为ph10.0系统适用性溶液图谱。图8是实施例8的hplc图谱,其中,图8a为a液:b液(v/v)=85:15时的系统适用性溶液图谱,图8b为a液:b液(v/v)=80:20时的系统适用性溶液图谱,图8c为a液:b液(v/v)=75:25时的系统适用性溶液图谱,图8d为a液:b液(v/v)=70:30时的系统适用性溶液图谱。图9是实施例9的hplc图谱,其中,图9a为流速0.8ml/min系统适用性溶液图谱,图9b为流速1.0ml/min系统适用性溶液图谱,图9c为流速1.2ml/min系统适用性溶液图谱。图10是实施例10的hplc图谱,其中,图10a为波长210nm系统适用性溶液图谱,图10b为波长225nm系统适用性溶液图谱,图10c为波长260nm系统适用性溶液图谱。图11是实施例11的hplc图谱,其中,图11a为进样量20μl系统适用性溶液图谱,图11b为进样量50μl系统适用性溶液图谱。图12是实施例12的hplc图谱,其中,图12a为碳八固定相系统适用性溶液图谱,图12b为碳十八固定相系统适用性溶液图谱。图13是对比例1的hplc图谱,其中,图13a为空白溶液,图13b为系统适用性溶液,图13c为富马酸福莫特原料药供试品溶液图谱。图14是对比例2的hplc图谱,其中,图14a为空白溶液,图14b为系统适用性溶液,图14c为富马酸福莫特罗吸入气雾剂供试品溶液图谱。图15是对比例3的hplc图谱,其中,图15a为供试品溶液0小时的色谱图,图15b为供试品溶液5小时的色谱图。具体实施例方式下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的药材原料、试剂材料等,如无特殊说明,均为市售购买产品。实施例及对比例中所用富马酸福莫特罗原料药和气雾剂如无特殊说明,富马酸福莫特罗原料药为新乡海滨药业有限公司生产;富马酸福莫特罗气雾剂为深圳太太药业有限公司生产。仪器:安捷伦高效液相色谱仪aglient1260(dad)高效液相色谱仪;色谱柱:碳十八烷基硅烷键合相色谱柱watersxbridgec18(250mm×4.6mm,5μm)、agilentzorbaxsb-c8(250mm×4.6mm,5μm)。实施例1检测条件:取碳十八烷基硅烷键合相色谱柱watersxbridgec18(250mm×4.6mm,5μm);a液为(0.6%,v/v)四丁基氢氧化铵水溶(磷酸调节ph=8.5),b液为乙腈,a液与b液体积比为80:20;柱温:35℃;流速:1.0ml/min;检测波长:225nm;进样体积:50μl。实验步骤:空白溶液(稀释液)的配制:分别量取乙腈120ml和水880ml,摇匀,即得。系统适用性溶液的配制:称取富马酸福莫特罗杂质i鉴别用对照品(edqm,欧洲官方对照品)约2mg,置20ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,即得。供试品溶液的配制:取富马酸福莫特罗气雾剂1瓶(规格:120揿/瓶,每揿12μg,厂家:深圳太太药业有限公司,批号180030-1),在室温下倒置(阀杆朝下)1小时,采用尖头锥在样品罐底部扎一小孔,逸去抛射剂后,用割管器除去样品罐底部,用稀释液将内容物溶解并转移至20ml量瓶中,加稀释液至刻度,摇匀,作为供试品溶液。测定:取空白溶液、系统适用性溶液与供试品溶液各进样一针。按上述条件进行高效液相色谱分析,记录色谱图,按峰面积归一化法计算富马酸福莫特罗杂质i含量。结果表明,该批次供试品的杂质i的含量为0.26%。hplc色谱图见附图1,其中,图1a为空白溶液,图1b为系统适用性溶液,图1c为富马酸福莫特罗吸入气雾剂供试品溶液图谱。实施例2检测条件:同实施例1。实验步骤:空白溶液(稀释液)、系统适用性溶液、供试品溶液的配制:同实施例1。杂质i定位溶液:称取富马酸福莫特罗杂质i对照品(edqm)约14mg,置20ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,即得。杂质a定位溶液:称取约1mg富马酸福莫特罗杂质a(来源:tlc公司)对照品置100ml量瓶中,加稀释液使溶解并稀释至刻度,摇匀,精密移取该溶液1ml至10ml量瓶,用稀释液定容至刻度,摇匀,即得。测定:取系统适用性溶液、供试品溶液、杂质i定位溶液、杂质a定位溶液各进样一针。按上述条件进行高效液相色谱分析,记录色谱图。由实验结果可以看出,富马酸福莫特罗主峰保留时间为25.420min、杂质a保留时间为19.027min、杂质i保留时间为29.020min;且色谱图基线平稳,主峰峰形良好,主峰与杂质峰、杂质与杂质之间能够实现了良好的分离。hplc色谱图见附图2,其中,图2a为杂质a定位溶液,图2b为杂质i定位溶液,图2c为富马酸福莫特罗吸入气雾剂供试品溶液图谱。其中:杂质a的结构为(杂质a为福莫特罗的降解杂质):实施例3检测条件:同实施例1。实验步骤:空白溶液(稀释液)、系统适用性溶液的配制:同实施例1。供试品溶液的配制:精密称取富马酸福莫特罗原料药7mg,置于100ml容量瓶中,加稀释液稀释溶解并稀释至刻度,摇匀,即得;测定:取空白溶液、系统适用性溶液及供试品溶液各进样一针。按上述条件进行高效液相色谱分析,记录色谱图,按峰面积归一化法计算富马酸福莫特罗杂质i含量。结果表 明,该批次供试品的杂质i的含量为0.14%,hplc色谱图见附图3,图3a为空白溶液图谱,图3b为系统适用性溶液图谱,图3c为富马酸福莫特罗原料药供试品溶液图谱。实施例4检测条件:同实施例1。实验步骤:稀释液、系统适用性溶液的配制:同实施例1。测定:取系统适用性溶液进样1针作为0小时系统适用性溶液,在色谱柱持续使用约40小时之后再进样一针作为40小时系统适用性溶液。按上述条件进行高效液相色谱分析,记录色谱图。结果为,40小时系统适用性溶液的杂质i与主峰之间的分离度为3.80,0小时系统适用性溶液的杂质i与主峰之间的分离度为3.79,结果表明,经过长时间的使用,色谱柱柱效无下降。hplc色谱图见附图4,其中,图4a为0小时系统适用性溶液,图4b为40小时系统适用性溶液。缓冲液离子浓度、ph值、检测波长、流动相比例等因素,都影响富马酸福莫特罗、非对映杂质以及其它杂质的分离。因此,通过下述实施例对色谱条件进行优选。实施例5柱温的选择检测条件:同实施例1。实验步骤:稀释液、系统适用性溶液的配制:同实施例1。测定:分别于柱温25℃、35℃、40℃、45℃、50℃考察柱温对杂质i检测的影响,检测结果见表1,hplc图谱见附图5。表1柱温对杂质i检测的影响由实验结果可以看出,当柱温分别为25℃、35℃、40℃、45℃、50℃,检测出的杂质i含量基本一致,主峰随着柱温的升高保留增强。其中以柱温为35℃与40℃时的分离效果最好,考虑到柱温低有助于提高色谱柱的使用寿命,因此,优选柱温为35℃。hplc色谱图见附图5,其中,图5a为25℃系统适用性溶液图谱,图5b为35℃系统适用性溶液图谱,图5c为40℃系统适用性溶液图谱,图5d为45℃系统适用性溶液图谱,图5e为50℃系统适用性溶液图谱。实施例6缓冲液离子浓度的选择检测条件:取碳十八烷基硅烷键合相固定相色谱柱watersxbridgec18(250mm×4.6mm,5μm);a液分别为0.3%、0.6%或0.7%(v/v)的四丁基氢氧化铵水溶(磷酸调节ph=8.5),b液为乙腈,a液与b液体积比为80:20;流速:1.0ml/min;检测波长:225nm;进样体积:50μl。系统适用性溶液的配制:同实施例1。测定:分别考察缓冲液不同离子浓度对杂质i检测的影响,检测结果见表2,hplc图谱见附图6。表2缓冲液不同离子浓度对杂质i检测的影响离子浓度(v/v)0.2%0.3%0.6%0.7%杂质i(%)0.230.320.340.32主峰保留时间(min)12.57321.04724.57331.793主峰与杂质i的分离度2.693.473.523.88由实验结果可以看出,缓冲液的离子浓度不同(0.3%~0.7%),检测出的杂质i含量基本一致,主峰保留时间、杂质i与主峰的分离度随着离子浓度的增加而增大。但是离子浓度降低至0.2%,主峰及杂质i保留明显降低,主峰与杂质i的分离度明显降低,另外考虑到离子对浓度的增加可能会影响色谱柱的使用寿命,因此,优选离子对浓度为0.6%(v/v)。hplc色谱图见附图6,其中,图6a为0.2%(v/v)系统适用性溶液图谱,图6b为0.3%(v/v)系统适用性溶液图谱,图6c为0.6%(v/v)系统适用性溶液图谱,图6d为0.7%(v/v)系统适用性溶液图谱。实施例7缓冲液ph值的选择检测条件:取碳十八烷基硅烷键合相固定相色谱柱watersxbridgec18(250mm×4.6mm,5μm);a液为0.6%四丁基氢氧化铵水溶液,分别用磷酸调节ph至7.0、8.5、10.0,b液为乙腈,a液与b液体积比为80:20;流速:1.0ml/min;检测波长:225nm;进样体积:50μl。系统适用性溶液的配制:同实施例1。测定:分别考察不同ph值缓冲液对杂质i检测的影响,检测结果见表3,hplc图谱见附图7。表3缓冲液不同ph值对杂质i检测的影响缓冲液ph值6.07.08.510.0杂质i(%)0.210.330.340.33主峰保留时间(min)11.05318.77324.57328.993主峰与杂质i的分离度2.343.213.524.41由实验结果可以看出,缓冲液的ph值不同(7.0~10.0),检测出的杂质i含量基本一致,主峰保留时间随着ph的上升而增大,杂质i与主峰的分离度也随着离子浓度的增加而增加,但是ph降低至6.0时,主峰及杂质i保留明显降低,主峰与杂质i的分离度明显降低,思考这可能与流动相此时成酸性有关,而杂质i与主成分在碱性条件中保留和分离能力较好。另外考虑到ph值的增加可能会影响色谱柱的使用寿命,因此,优选缓冲液ph值为8.5。hplc色谱图见附图7,其中,图7a为ph6.0系统适用性溶液图谱,图7b为ph7.0系统适用性溶液图谱,图7c为ph8.5系统适用性溶液图谱,图7d为ph10.0系统适用性溶液图谱。实施例8流动相比例的选择检测条件:取碳十八烷基硅烷键合相固定相色谱柱watersxbridgec18(250mm×4.6mm,5μm);a液为0.6%(v/v)四丁基氢氧化铵水溶液(用磷酸调节ph至8.5),b液为乙腈,a液与b液体积比分别为85:15或80:20或75:25;流速:1.0ml/min;检测波长:225nm;进样体积:50μl。系统适用性溶液的配制:同实施例1。测定:分别采用不同的洗脱比例,考察洗脱比例对杂质i检测的影响,检测结果见表4,hplc图谱见附图8。表4不同的洗脱比例对杂质i检测的影响a液:b液(v/v)85:1580:2075:2570:30杂质i(%)0.340.340.310.19%主峰保留时间(min)25.20724.57311.12010.313主峰与杂质i的分离度3.713.523.112.25由实验结果可以看出,流动相比例不同(a液:b液(v/v)=(85:15)~(75:25)),检测出的杂质i含量基本一致,随着流动相乙腈比例的增加,主峰保留时间减小,杂质i与主峰的分离度降低,当流动相乙腈比例的增加至30%时,杂质i与主峰的分离度降低至2.25,杂质i的含量也相应的减小。主峰的保留时间随乙腈比例的增大而减小,较小或者较大的保留时间都不利于检测数据的分析,因此,优选a液:b液(v/v)=85:20。hplc色谱图见附图8,其中,图8a为a液:b液(v/v)=85:15时的系统适用性溶液图谱;图8b为a液:b液(v/v)=80:20时的系统适用性溶液图谱;图8c为a液:b液(v/v)=75:25时的系统适用性溶液图谱;图8d为a液:b液(v/v)=70:30时的系统适用性溶液图谱。实施例9流速的选择检测条件:取碳十八烷基硅烷键合相固定相色谱柱watersxbridgec18(250mm×4.6mm,5μm);a液为0.6%(v/v)四丁基氢氧化铵水溶液(用磷酸调节ph至8.5),b液为乙腈,a液与b液体积比为80:20;流速分别为:0.8或1.0或1.2ml/min;检测波长:225nm;进样体积:50μl。系统适用性溶液的配制:同实施例1。测定:分别于考察缓冲液不同离子浓度对杂质i检测的影响,检测结果见表5,hplc图谱见附图9。表5不同流速对杂质i检测的影响由实验结果可以看出,流动相乙腈比例不同,检测出的杂质i含量基本一致,杂质i与主峰的分离度没有明显差异,主峰保留时间随流速的增加而降低,但较大或较小的保留时间都会对检测结果的分析产生不利影响,因此优选流速为1.0。hplc色谱图见附图9,其中,图9a为流速0.8ml/min系统适用性溶液图谱,图9b为流速1.0ml/min系统适用性溶液图谱,图9c为流速1.2ml/min系统适用性溶液图谱。实施例10波长的选择检测条件:取碳十八烷基硅烷键合相固定相色谱柱watersxbridgec18(250mm×4.6mm,5μm);a液为0.6%(v/v)四丁基氢氧化铵水溶液(用磷酸调节ph至8.5),b液为乙腈,a液与b液体积比为80:20;流速:1.0ml/min;检测波长分别为:210nm、225nm、260nm;进样体积:50μl。系统适用性溶液的配制:同实施例1。测定:进样系统适用性溶液,考察检测波长不同对杂质i检测的影响,检测结果见表6,hplc图谱见附图10。表6不同波长对杂质i检测的影响波长(nm)210225260杂质i(%)0.340.340.36主峰保留时间(min)24.57324.57324.573主峰与杂质i的分离度3.593.523.48由实验结果可以看出,检测波长不同,检测出的杂质i含量基本一致,杂质i与主峰的分离度没有明显差异,但由于福莫特罗及相关杂质的紫外吸收光谱差别较大,较小或者较大的波长对检测结果均会产生不利影响,综合考虑,优选波长为225nm。hplc色谱图见附图10,其中,图10a为波长210nm系统适用性溶液图谱,图10b为波长225nm系统适用性溶液图谱,图10c为波长260nm系统适用性溶液图谱。实施例11进样量的选择检测条件:取碳十八烷基硅烷键合相固定相色谱柱watersxbridgec18(250mm×4.6mm,5μm);a液为0.6%(v/v)四丁基氢氧化铵水溶液(用磷酸调节ph至8.5),b液为乙腈,a液与b液体积比为80:20;流速:1.0ml/min;检测波长:225nm;进样体积分别为:20μl或50μl。系统适用性溶液的配制:同实施例1。测定:分别采用进样量为20μl或50μl进行检测,考察不同进样量对杂质i检测的影响,检测结果见表7,hplc图谱见附图11。表7不同进样量对杂质i检测的影响进样量(μl)2050杂质i(%)0.350.34主峰保留时间(min)24.19324.573主峰与杂质i的分离度4.453.52由实验结果可以看出,进样量不同,检测出的杂质i含量基本一致,虽然较大的进样量会导致分离度降低,但是低进样量会降低检测的灵敏度,综合考虑,优选流速为50μl。hplc色谱图见附图11,其中,图11a进样量为20μl系统适用性溶液图谱,图11b进样量为50μl系统适用性溶液图谱。实施例12色谱柱固定相的选择检测条件:取碳十八烷基硅烷键合相固定相色谱柱watersxbridgec18(250mm×4.6mm,5μm);或碳八烷基硅烷键合相固定相色谱柱agilentzorbaxsb-c8(250mm×4.6mm,5μm);流动相:a液为a液为0.6%四丁基氢氧化铵水溶液(用磷酸调节ph至8.5),b液为乙腈,a液与b液体积比为80:20;流速:1.0ml/min;检测波长:225nm;进样体积:50μl。系统适用性溶液的配制:同实施例1。测定:分别于考察缓冲液不同离子浓度对杂质i检测的影响,检测结果见表8,hplc图谱见附图12。表8不同固定相种类对杂质i检测的影响固定相种类碳八碳十八杂质i(%)0.320.34主峰保留时间(min)21.40024.573主峰与杂质i的分离度3.213.52由实验结果可以看出,固定相不同,检测出的杂质i含量相同,而采用碳十八烷基硅烷键合相固定相色谱柱watersxbridgec18(250mm×4.6mm,5μm)的分离度更高,保留时间合理,因此优选碳十八烷基硅烷键合相为固定相。hplc色谱图见附图12,其中,图12a为碳八烷基硅烷键合相固定相系统适用性溶液图谱,图12b为碳十八烷基硅烷键合相固定相系统适用性溶液图谱。对比例1参照中国药典2015版富马酸福莫特罗非对映异构体检测方法:检测条件:取十八烷基键合聚乙烯醇色谱柱shodexasahipakodp-504e(250mm×4.6mm,5μm);以磷酸盐缓冲液(取磷酸钾三水合物5.3g,加水1000ml溶液,用5mol/l氢氧化钾溶液或磷酸调节ph值至12.0±0.1)-乙腈(88:12)为流动相;柱温:30℃;流速:0.5ml/min;检测波长:225nm;进样体积:20μl。实验步骤:空白溶液(稀释液)的配制:分别量取乙腈120ml和水880ml,摇匀,即得。系统适用性溶液的配制:称取富马酸福莫特罗杂质i鉴别用对照品约2mg,置20ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,即得。供试品溶液的配制:精密称取富马酸福莫特罗原料药7mg,置于100ml容量瓶中,加稀释液稀释溶解并稀释至刻度,摇匀,即得。测定:取空白溶液、系统适用性溶液与供试品溶液进样一针,按上述条件进行高效液相色谱分析,记录色谱图,按峰面积归一化法计算富马酸福莫特罗杂质i含量。结果表明该批次供试品的杂质i的含量为0.13%。hplc色谱图见附图13,其中,图13a为空白溶液,图13b为系统适用性溶液,图13c为富马酸福莫特原料药供试品溶液图谱。对比例2参照中国药典2015版富马酸福莫特罗非对映异构体检测方法:检测条件:取十八烷基键合聚乙烯醇色谱柱shodexasahipakodp-504e(250mm×4.6mm,5μm);以磷酸盐缓冲液(取磷酸钾三水合物5.3g,加水1000ml溶液,用5mol/l氢氧化钾溶液或磷酸调节ph值至12.0±0.1)-乙腈(88:12)为流动相;柱温:30℃;流速:0.5ml/min;检测波长:225nm;进样体积:20μl。实验步骤:空白溶液(稀释液)、系统适用性溶液、供试品溶液的配制:同实施例1。测定:取空白溶液、系统适用性溶液与供试品溶液进样一针。按上述条件进行高效液相色谱分析,记录色谱图,按峰面积归一化法计算富马酸福莫特罗杂质i含量。结果表明该批次供试品的杂质i的含量为0.24%。hplc色谱图见附图14,其中,图14a为空白溶液,图14b为系统适用性溶液,图14c为富马酸福莫特罗吸入气雾剂供试品溶液图谱。对比例3检测条件:同对比例1;实验步骤:供试品溶液的配制:同实施例1。测定:取供试品溶液立即进样1针作为0小时的色谱图,色谱柱持续使用5小时之后再进样1针作为5小时的色谱图。结果表明,0小时供试品溶液的杂质i与主峰分离度为3.20,经过5小时的使用,杂质i与主峰分离度为2.00,色谱柱柱效严重下降。hplc色谱图见附图15,其中,图15a为供试品溶液0小时的色谱图,图15b为供试品溶液5小时的色谱图。由实施例3与对比例1、实施例1与对比例2的检测结果可以看出,采用本发明所述方法与参照中国药典记载的方法检测富马酸福莫特罗原料药及其相关制剂中非对映异构体的结果一致;而由实施例4与对比例3检测结果可以看出,本发明所述方法色谱柱耐受性更好,经过长时间使用,柱效无明显下降。综上所述,本发明所述的检测方法,色谱柱耐受性好、使用寿命长,检测结果准确、重现性好,并能同时适用于检测富马酸福莫特罗原料药及其相关制剂中非对映异构体杂质的含量。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1