一种测定发酵豆粕中乳酸含量的方法以及测定用待测液的提取方法与流程
本发明涉及发酵豆粕中乳酸含量的测定方法领域,具体而言,涉及一种测定发酵豆粕中乳酸含量的方法以及测定用待测液的提取方法。
背景技术:
发酵豆粕是以优质豆粕为主要原料,在多菌种混合发酵下,将豆粕中的多种抗营养因子去除,同时其在发酵过程中产生大量对动物有益的菌种、乳酸等物质。豆粕在发酵过程中产生多种有机酸,乳酸是其中最多的一种有机酸。
目前测定发酵豆粕中乳酸含量的方法基本上都是碱滴定法或高效液相色谱法。其中,碱滴定法存在很多问题,滴定的结果反应的是总酸的含量,无法准确确定乳酸的量。而高效液相法由于发酵豆粕中成分复杂,杂峰干扰大,也无法准确进行测定。
在专利文献cn103048403a-一种发酵豆粕中乳酸的测定方法中,记载了通过80%的乙醇将发酵豆粕中蛋白质类物质沉淀下来并超声波提取乳酸,上清液再用0.45μm滤膜过滤去除大分子物质,采用甲醇和盐酸混合流动相分离发酵豆粕中的乳酸,外标法计算乳酸含量。
该专利方法虽然测量过程较为快速,但因发酵豆粕中成分复杂,若直接采用80%乙醇溶液在对乳酸进行超声提取的过程中对蛋白质类物质进行沉淀,则会导致最后测量精度与准确度不高。
鉴于此,特提出本申请。
技术实现要素:
本发明的目的在于提供一种测定发酵豆粕中乳酸含量的方法,其利用高效液相色谱对特定的待测液进行检测,其能保证了乳酸提取充分的同时确保色谱基线的平稳性,检测结果分辨率稳定、误差小、准确性佳。
本发明的另一目的在于提供一种测定用待测液的提取方法,其对乳酸提取更为充分,待测液中杂质含量低,能够提高后续乳酸含量测定的结果准确性。
本发明的实施例是这样实现的:
一种测定发酵豆粕中乳酸含量的方法,其包括将待测液进行高效液相色谱检测;其中,所述待测液的提取方法包括:用水提取样品,提取后分离,取第一上清液,向所述第一上清液中加入无水乙醇以沉淀蛋白,静置后分离,取第二上清液作为待测液。
一种测定用待测液的提取方法,其包括:用水提取样品,提取后分离,取第一上清液,向所述第一上清液中加入无水乙醇以沉淀蛋白,静置后分离,取第二上清液作为待测液。
本发明实施例的有益效果例如包括:
本申请在进行高效液相色谱检测之前,通过对样品进行水提,能够充分、均匀得提取出发酵豆粕中的乳酸,同时能够避免因提取试剂选择不合适而影响乳酸提取效果的问题,确保了乳酸的充分提取。随后对蛋白进行沉淀,本申请选用先水提后沉淀的方式进行,能够避免无水乙醇用量过大,导致乙醇体积浓度过大而造成溶液中出现共沉现象,或出现蛋白质局部变性的情况,进而导致蛋白质沉淀效果欠佳,获得的待测液体积浓度在乳酸标准品曲线范围内,乳酸提取充分,蛋白质经有效沉淀后,第二上清液经分离能够分离掉其他杂质分子,本申请在利用高效液相色谱对该待测液进行检测时,能够有效去除杂峰干扰,保证了乳酸提取充分的同时确保色谱基线的平稳性,并且分辨率稳定、误差小,提高了测定精度与准确性,本方法测得的数据稳定性好,更具有可靠性,适合推广。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
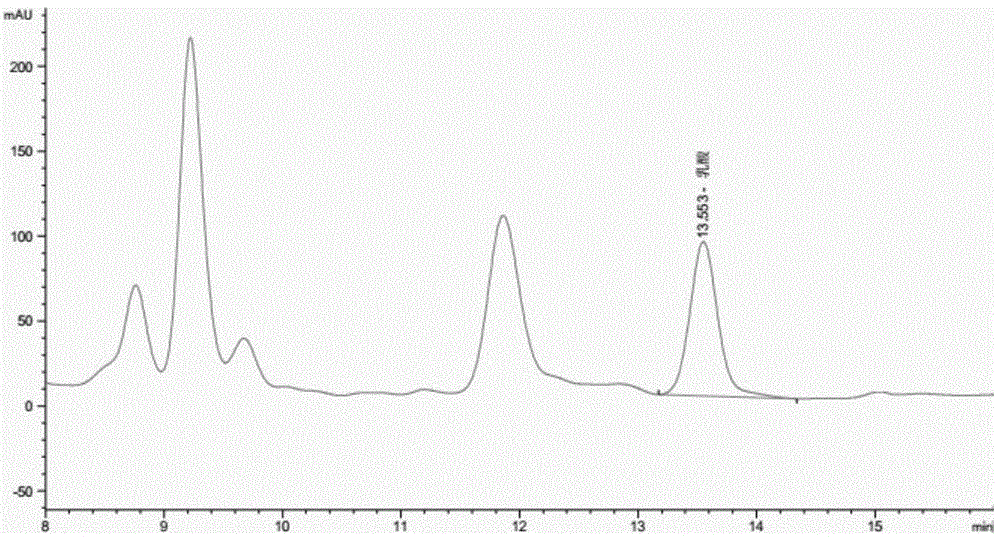
图1为实施例1提供的色谱图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
下面对本发明实施例的测定发酵豆粕中乳酸含量的方法以及测定用待测液的提取方法进行具体说明。
本申请提供了一种测定发酵豆粕中乳酸含量的方法,其包括以下步骤:
s1、提取乳酸
用水提取样品,提取后分离,取第一上清液。
乳酸能够与水混溶,本申请中对样品中的乳酸进行水提,并且于水浴条件下进行提取,提取较为完全。
其中,样品与水的料液比为1g:18-22ml;优选为1g:19-21ml。提取的温度为50-70℃,例如可以为50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69以及70℃中的任一者或者任意两者之间的范围值,优选为60℃;利用水提取样品的提取时间为1-1.5h。
在本实施例中,提取是在振荡条件下进行的,优选地,是在水浴摇床中进行,更具体地,在提取时水浴摇床的转速(以旋转的方式)为100-130r/min。
水包括但不限于超纯水、去离子水、纯水中的任一者,优选地,本申请中水为超纯水。
提取后分离包括利用滤纸过滤或于8000-12000r/min下离心过滤8-12min。其中,如果采用滤纸进行过滤时,由于采用干过滤法,滤液在经过滤纸时,滤纸会吸收部分水分,所以初滤液的实际体积浓度要高于其原体积浓度,因此,在过滤时,需要弃去初滤液。
同时,初滤液可以起到润洗承接容器的作用,在才刚刚开始过滤时,滤纸孔隙大,经过一段过滤后,滤纸孔被附着的滤渣所覆盖,孔隙变小,滤液也会更加澄清。进一步地,滤纸本身加工不那么的精细,表面有一些可溶性的杂质,本申请中通过除去初滤液能够有效防止这些杂质进入溶液当中,有效去除杂峰干扰,保证溶液的纯度,并且不会影响乳酸含量的测定。
s2、沉淀蛋白
向第一上清液中加入无水乙醇以沉淀蛋白,静置后分离,取第二上清液。
第一上清液与无水乙醇的体积比为2-9:1-8;优选为5-9:1-5。经发明人研究发现,随着无水乙醇的体积增大,对应的溶液中乙醇体积浓度增加,会导致溶液中的蛋白无法达到预期的沉淀效果。这是因为,无水乙醇体积过大,会使溶液的总体积加大,也起到了稀释溶液中乳酸含量的作用,鉴于发酵豆粕中乳酸含量较低,进一步被稀释后会导致沉淀效果不佳。经发明人研究还发现,高体积浓度的乙醇(>70%)会导致蛋白质变性,或者是快速加入无水乙醇时,也会导致溶液中局部乙醇体积浓度过高,也会导致蛋白质变性,进而导致无法沉淀其他蛋白质,沉淀效果欠佳。此外,乙醇的体积浓度过高会导致后续的色谱分析时影响乳酸的分离。
因此,本申请中限定第一上清液与无水乙醇的体积比为5-9:1-5,此时第一上清液和无水乙醇混合而成的溶液中乙醇的体积浓度小于等于50%,此时,溶液中的蛋白质能够很好的被沉淀,同时能够有效避免高体积浓度乙醇导致蛋白质变性或在色谱分析时影响乳酸分离的情况发生。
进一步地,本申请中,在加入无水乙醇时,缓慢加入,并且采用边加边摇动的方式加入,能够有效避免无水乙醇加入过快而导致溶液局部乙醇含量过高的情况产生。
此外,还值得注意的是,发酵豆粕中成分较多,包括了各种蛋白质、寡糖、小肽、有机酸和异黄酮等,其中蛋白质有多种,例如:纤维蛋白原、球蛋白以及白蛋白等等,经发明人研究发现,6-10%的乙醇溶液更易沉淀纤维蛋白原,18-22%的乙醇溶液更容易沉淀球蛋白,而38-42%的乙醇溶液更容易沉淀白蛋白。本申请中采用缓慢加入无水乙醇的方式能够使得溶液中乙醇的含量逐步加大,从而在乙醇溶液体积浓度逐步加大的过程中逐步对各种蛋白质进行沉淀,并且在加入全部的无水乙醇后,通过静置20-30min的方式使得蛋白质的沉淀更完全。
本申请中,通过控制加入无水乙醇的速度,能够实现缓慢均匀的加入,相较于直接快速加入所有无水乙醇而言,浓度一次性达到较大浓度,此时容易造成溶液发生共沉现象,同时还有可能造成蛋白质局部变性,无法正常沉淀。本申请通过缓慢加入的方式逐渐增加浓度,使得各个蛋白质在加入无水乙醇的过程中进行沉淀,随后静置能够保证之前加入过程中未沉淀完全的蛋白继续沉淀,充分降低溶液中蛋白的含量,保证色谱曲线的平稳性。
s3、微孔滤膜过滤
将第二上清液过0.22μm滤膜,0.22μm的滤膜可以达到gmp或者药典规定的除菌99.99%的要求,对其他杂质分子的去除效果更佳,而0.45μm的滤膜主要去除大分子物质,例如颗粒和大多数细菌微生物,本申请选用0.22μm的滤膜的过滤效果更佳。
s4、高效液相色谱分析
对步骤s3获得的待测液进行高效液相色谱检测。
采用高效液相色谱进行检测时的检测波长为200-220nm;
优选地,采用高效液相色谱进行检测时的色谱柱为hi-plexh、流动相为0.1%-0.2%三氟乙酸、流速为0.5-0.7ml/min、柱温为42-48℃、进样量为10-20μl。
由于本申请中发酵豆粕中的乳酸提取充分,获得的待测液线性关系好,能够提高测定精度与准确性,因此,本申请中选择了特定的色谱柱(hi-plexh)、并且对应的选用了较高的柱温,例如42-48℃,同时选用较低的流速0.5-0.7ml/min,进行色谱分析。
通常情况下,降低流动相的流速可以提高柱效,但分析时间相对加长,现有技术中流速通常为0.9-1.1ml/min。而适当提高色谱柱温度,可以降低流动相粘度,提高组分传质速度,提高分析速度,但是同时会降低分离度,所以现有技术中柱温通常为25-35℃。
相较于现有技术,本申请中的柱温明显增高,流速明显降低,本申请采用升高柱温,降低流速的方式来平衡分离过程,同时达到更好的柱效,上述柱温和流速与本申请中特定的色谱柱是相适应进行选择的,经发明人研究发现,采用上述特定的色谱条件能够更好的对乳酸进行分离,能够保证样品的保留时间稳定,分辨率稳定,且整个测定过程不仅仅保证了乳酸提取充分,同时确保液相图谱的基线更加平稳,并且分辨率稳定、误差小,同时还能保证测定精度与准确性。
在确定了色谱条件后,按照常规的方法绘制峰面积或峰高的标准曲线,然后对待测液按照上述色谱条件进行检测,测定色谱峰面积或峰高,与标准曲线进行比较以确定待测液中乳酸的质量。
以下结合实施例对本发明的测定发酵豆粕中乳酸含量的方法以及测定用待测液的提取方法进一步进行阐述。
实施例1
本实施例提供了一种测定用待测液的提取方法,其包括以下步骤:
精确称量样品约5.0g,加入100ml超纯水,在60℃水浴摇床提取1h后,水浴摇床的转速为115r/min,取出,冷却至室温,滤纸过滤(弃去初滤液)。取第一上清液14ml,以1ml/min的加入速度缓慢均匀地加入(边加边摇)无水乙醇6ml(此时,第一上清液与无水乙醇的体积比为7:3),静置20min,滤纸过滤。取第二上清液过0.22μm滤膜作为待测液。
实施例2
本实施例提供了一种测定用待测液的提取方法,其包括以下步骤:
精确称量样品约5g,加入90ml超纯水,在53℃水浴摇床提取1.4h后,取出,冷却至室温,摇匀,10000r/min离心10min。取第一上清液14ml,缓慢均匀地加入(边加边摇)无水乙醇6ml,静置20min,离心过滤。取第二上清液过0.22μm滤膜作为待测液。
实施例3
本实施例提供了一种测定用待测液的提取方法,其包括以下步骤:
精确称量样品约5g,加入95ml超纯水,在62℃水浴摇床提取1.2h后,取出,冷却至室温,摇匀,9000r/min离心12min。取第一上清液12ml,缓慢均匀地加入(边加边摇)无水乙醇8ml,接着静置28min,离心过滤。取第二上清液过0.22μm滤膜作为待测液。
实施例4
本实施例提供了一种测定用待测液的提取方法,其包括以下步骤:
精确称量样品约5g,加入105ml超纯水,在70℃水浴摇床提取1h后,取出,冷却至室温,摇匀,12000r/min离心18min。取第一上清液10ml,以1ml/min的加入速度缓慢均匀地加入(边加边摇)无水乙醇10ml,静置23min,离心过滤。取第二上清液过0.22μm滤膜作为待测液。
实施例5
本实施例提供了一种测定用待测液的提取方法,其包括以下步骤:
精确称量样品约5g,加入110ml超纯水,在65℃水浴摇床提取1h后,取出,冷却至室温,摇匀,11000r/min离心9min。取第一上清液10ml,以0.8ml/min的加入速度缓慢均匀地加入(边加边摇)无水乙醇10ml,静置25min,离心过滤。取第二上清液过0.22μm滤膜作为待测液。
实施例6
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:本实施例中,取第一上清液18ml,加入无水乙醇2ml(此时,第一上清液与无水乙醇的体积比为9:1)。
实施例7
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:本实施例中,取第一上清液16ml,加入无水乙醇4ml(此时,第一上清液与无水乙醇的体积比为8:2)。
实施例8
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:本实施例中,取第一上清液12ml,加入无水乙醇8ml(此时,第一上清液与无水乙醇的体积比为6:4)。
实施例9
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:本实施例中,取第一上清液10ml,加入无水乙醇10ml(此时,第一上清液与无水乙醇的体积比为5:5)。
实施例10
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:本实施例中,取第一上清液8ml,加入无水乙醇12ml(此时,第一上清液与无水乙醇的体积比为4:6)。
实施例11
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:本实施例中,取第一上清液6ml,加入无水乙醇14ml(此时,第一上清液与无水乙醇的体积比为3:7)。
实施例12
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:本实施例中,取第一上清液4ml,加入无水乙醇16ml(此时,第一上清液与无水乙醇的体积比为2:8)。
对比例1
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:取第一上清液20ml,此时,不加入无水乙醇。
对比例2
本实施例提供的测定用待测液的提取方法与实施例1基本相同,区别在于:样品和纯水的料液比为1:25g/ml,即向5g样品中加入125ml的纯水。
对比例3
采用专利文献cn103048403a中的提取方法来提取:
精确称量样品约5.0g,置于100ml容量瓶中,加入80%的乙醇溶液定容至100ml,超声波50℃提取30min,冷却至室温,样品液用高速冷冻离心机离心,转速12000rpm,4℃离心10min,上清液过0.45μm滤膜,作为待测液。
对比例4
按照国标gb/t12456-2008食品中总酸的测定方法来进行测定,通过酸碱滴定法(人工判断滴定终点,通过酸的换算系数计算乳酸含量)来进行测定。测得其乳酸含量为1.40%。
实施例13
本实施例提供了一种测定发酵豆粕中乳酸含量的方法,上述实施例1-12以及对比例1-3均是采用本实施例提供的测定方法进行检测,具体是分别取实施例1-12中制备的待测液10μl进行hplc分析,其中,色谱条件包括:
仪器设备:agilent1260,配备紫外检测器,检测波长210nm;
色谱柱:hi-plexh(7.7mm×300mm,8μm)
流动相:0.1%三氟乙酸
柱温:45℃;
流速:0.6ml/min;
进样量:10μl。
首先,按照色谱条件绘制标准曲线,绘制标准曲线包括:分别取乳酸标准液5μl、10μl、15μl、20μl以及25μl(相当于乳酸含量25μg、50μg、75μg、100μg以及125μg)注入液相色谱仪中,以上述实施例1-12以及对比例1-3获得的待测液进行高效液相色谱分析,测定色谱峰面积,以标准乳酸质量对相应的峰面积做标准曲线(标准曲线为y=1.569x+7.257,r2=1.0000)。
其中,实施例1按照实施例13提供的测定方法进行检测获得的色谱图请参阅图1。将上述实施例1-12以及对比例1-3获得的峰面积,与对应的标准曲线进行比较以获得实施例1-12以及对比例1-3的待测液中乳酸的含量,以及对比例4的乳酸含量。检测结果请参阅表1:
表1.不同待测液在相同检测条件下的色谱测定结果
从上表可以看出,本申请实施例的效果明显优于对比例3的效果,由此说明,由此说明实施例提取效果更好。此外,从对比例1和对比例2可以看出,当料液比为1:20时,测定的乳酸含量略高于料液比为1:25时的测定结果,将实施例1和实施例7-12以及对比例1-2结合比较可以看出,加入适量无水乙醇,使得提取液中无水乙醇浓度≤50%会取得更好的实验效果,而不加入无水乙醇(对比例3)和加入无水乙醇的量大于50%后,效果明显欠佳。实施例1的测定结果要明显高于对比例4,由此说明实施例通过高效液相色谱仪测定精度与准确性好,并且分辨率稳定、误差小,避免因主观判断滴定终点带来的误差。
综上所述,本申请在进行高效液相色谱检测之前,通过在水浴条件下对样品进行水提,能够充分、均匀得提取出发酵豆粕中的乳酸,同时能够避免因提取试剂选择不合适而影响乳酸提取效果的问题,确保了乳酸的充分提取。随后对蛋白进行沉淀,本申请选用先水提后沉淀的方式进行,能够避免无水乙醇用量过大,导致乙醇体积浓度过大而造成溶液中出现共沉现象,或出现蛋白质局部变性的情况,进而导致蛋白质沉淀效果欠佳,获得的待测液体积浓度在乳酸标准品曲线范围内,乳酸提取充分,蛋白质经有效沉淀后,第二上清液经过滤能够过滤掉其他杂质分子,本申请在利用高效液相色谱对该待测液进行检测时,能够有效去除杂峰干扰,保证了乳酸提取充分的同时确保色谱基线的平稳性,并且分辨率稳定、误差小,提高了测定精度与准确性,本方法测得的数据稳定性好,更具有可靠性,适合推广。
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!