一种中药复方制剂指纹图谱的建立方法及其指纹图谱与流程
本发明涉及药物分析领域,尤其涉及一种中药复方制剂指纹图谱的建立方法及其指纹图谱。
背景技术:
治疗胃癌及其他消化道肿瘤的中药复方制剂为干蟾皮、壁虎、金荞麦、喜树果、西洋参、仙鹤草和灵芝的复方制剂,其中干蟾皮、壁虎(守宫)具消癥散结,破瘀止痛之功,为君药;金荞麦、喜树果攻削癌瘤,解毒散结,破血化瘀,为臣药;西洋参、仙鹤草、灵芝益气阴,安神志,养五脏,调和干蟾皮、守宫之毒性。君药、臣药、佐药、使药相伍,共奏消癥解毒,益气活血,化瘀止痛之效,使正气得存,邪不能犯,符合肿瘤病人邪盛正虚的病机特点,体现了“大霸微补,活血调气”的肿瘤治疗原则。临床主要协同治疗胃癌及其他消化道肿瘤证属血瘀者,以及用于进展期胃癌化疗后的协同治疗。
中药指纹图谱是近年来用于控制中药复方及其制剂质量的有效方法,中药指纹图谱技术为中药复方的质量控制研究提供了更广阔的视野。中药指纹图谱具有整体性的特点,突出中药的完整面貌。同时同类药材指纹图谱具有相似性的特点,依靠该图谱实现了对中药内在化学成分的综合评价和整体质量的全面控制,使中药制剂的成分更加可控。
hplc指纹图谱是目前应用最广泛的色谱分析法。hplc法是检测中药中各种化学成分的一种普遍适用的分析方法,其以高压力、高灵敏度、高效率以及自动化等优点广泛应用于中药化学成分检测中,使中药制剂的成分更加可控。
技术实现要素:
有鉴于此,本发明提供了一种中药复方制剂指纹图谱的建立方法及其指纹图谱,本发明提供的指纹图谱能够全面反映中药复方制剂的成分。
本发明提供了一种中药复方制剂指纹图谱的建立方法,其特征在于,包括以下步骤:
(1)采用甲醇对中药复方制剂进行超声提取,得到供试品溶液;
(2)采用高效液相色谱法对供试品溶液进行测试,根据《中药色谱指纹图谱相似度评价系统2012a版》,由得到的色谱数据生成中药复方制剂指纹图谱;
所述高效液相色谱法的色谱条件为:
色谱柱:c18色谱柱;
检测波长:200~220nm;
流动相:流动相a为体积分数0.05%~0.3%的磷酸水溶液,流动相b为乙腈;
洗脱方式为梯度洗脱,所述梯度洗脱的洗脱程序如表1所示。
优选的,所述中药复方制剂包括干蟾皮、壁虎、金荞麦、喜树果、西洋参、仙鹤草和灵芝;所述干蟾皮、壁虎、金荞麦、喜树果、西洋参、仙鹤草和灵芝的质量比为1~9:1~9:30~50:20~40:6~16:10~30:6~24。
优选的,所述色谱条件还包括:
流速:0.5~1.5ml/min;
柱温:15~35℃;
色谱柱规格:250mm×4.6mm,5μm;
进样体积:5~20μl;
进样浓度:0.01g/ml~1.00g/ml。
优选的,所述供试品溶液的制备方法包括以下步骤:
(a)将中药复方制剂与甲醇混合后超声,然后过滤,滤液减压浓缩,得到浓缩液;所述超声的功率为200~400w,频率为30~50khz,时间为20~60min;
(b)采用甲醇水溶液将浓缩液进行定容,然后将定容液中的上清液进行微孔滤膜过滤,得到供试品溶液。
优选的,所述步骤(b)中甲醇水溶液的体积浓度为10%~100%。
优选的,所述步骤(b)中微孔滤膜的孔径为0.2~0.5μm。
优选的,还包括配制混合对照品溶液,所述混合对照品溶液的配制方法包括以下步骤:将人参皂苷re、人参皂苷rg1、喜树碱、人参皂苷rb1、表儿茶素和蟾蜍噻咛对照品溶于甲醇中,得到混合对照品溶液;
采用与供试品溶液相同的高效液相色谱条件对混合对照品溶液进行测试,得到混合对照品溶液的色谱图;将混合对照品溶液的色谱图与权利要求1得到的中药复方制剂指纹图谱进行对比,用于指认中药复方制剂指纹图谱中的色谱峰。
本发明还提供了上述技术方案所述方法得到的中药复方制剂指纹图谱,以人参皂苷rb1色谱峰为参照,所述中药复方制剂指纹图谱包括29个共有峰,具体如下:
1号峰,相对保留时间为0.064;2号峰,相对保留时间为0.079;
3号峰,相对保留时间为0.088;4号峰,相对保留时间为0.097;
5号峰,相对保留时间为0.108;6号峰,相对保留时间为0.119;
7号峰,相对保留时间为0.134;8号峰,相对保留时间为0.143;
9号峰,相对保留时间为0.160;10号峰,相对保留时间为0.282;
11号峰,相对保留时间为0.294;12号峰,相对保留时间为0.311;
13号峰,相对保留时间为0.341;14号峰,相对保留时间为0.352;
15号峰,相对保留时间为0.411;16号峰,相对保留时间为0.436;
17号峰,相对保留时间为0.520;18号峰,相对保留时间为0.547;
19号峰,相对保留时间为0.570;20号峰,相对保留时间为0.639;
21号峰,相对保留时间为0.667;22号峰,相对保留时间为0.687;
23号峰,相对保留时间为0.898;24号峰,相对保留时间为0.929;
25号峰,相对保留时间为0.939;26号峰,相对保留时间为0.990;
27号峰,相对保留时间为1;28号峰,相对保留时间为1.058;
29号峰,相对保留时间为1.169。
优选的,以人参皂苷rb1色谱峰为参照,指纹图谱中的13号峰为蟾蜍噻咛;17号峰为表儿茶素;23号峰为人参皂苷re和rg1;24号峰为喜树碱;27号峰为人参皂苷rb1。
与现有技术相比,本发明提供的中药复方制剂指纹图谱的建立方法具有准确度高、稳定性高、重复性好的特点;本发明通过对11个不同批次的中药复方制剂进行检测,结果表明,相同位置色谱峰的相对保留时间的rsd均在5%之内,说明本发明提供的指纹图谱具有较好的重现性,本发明提供的中药复方制剂指纹图谱的建立方法可靠性较高;而且本发明由上述方法建立的中药复方制剂指纹图谱,具有29个达到有效分离的共有峰,所述指纹图谱能够有效地表征中药复方制剂的质量,有利于全面监控药物的质量。
附图说明
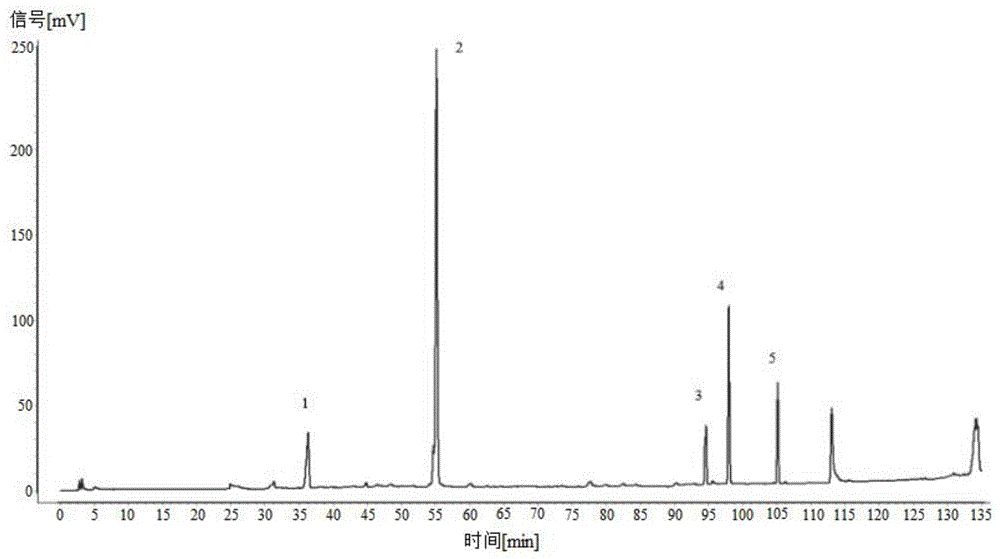
图1为本发明实施例4混合对照品的高效液相色谱图;
图2为本发明实施例4中11个批次中药复方制剂指纹图谱的叠加图谱;
图3为本发明实施例4中以11批供试品图谱为基础生成的对照图谱;
图4为本发明实施例4中各共有峰归属色谱图;
图5为本发明实施例4和对比例1不同波长色谱图;
图6为本发明实施例4和对比例2不同洗脱剂色谱图;
图7为本发明实施例4和对比例3不同提取溶剂色谱图。
具体实施方式
本发明提供了一种中药复方制剂指纹图谱的建立方法,包括以下步骤:
(1)采用甲醇对中药复方制剂进行超声提取,得到供试品溶液;
(2)采用高效液相色谱法对供试品溶液进行测试,根据《中药色谱指纹图谱相似度评价系统2012a版》,由得到的色谱数据生成中药复方制剂指纹图谱;
所述高效液相色谱法的色谱条件为:
色谱柱:c18色谱柱;
检测波长:200~220nm;
流动相:流动相a为体积分数0.05%~0.3%的磷酸水溶液,流动相b为乙腈;
洗脱方式为梯度洗脱,所述梯度洗脱的洗脱程序如表1所示。
本发明采用甲醇对中药复方制剂进行超声提取,得到供试品溶液。
在本发明中,所述中药复方制剂优选包括干蟾皮、壁虎、金荞麦、喜树果、西洋参、仙鹤草和灵芝;所述干蟾皮、壁虎、金荞麦、喜树果、西洋参、仙鹤草和灵芝的质量比优选为1~9:1~9:30~50:20~40:6~16:10~30:6~24。
在本发明中,所述供试品溶液的制备方法优选包括以下步骤:
(a)将中药复方制剂与甲醇混合后超声,然后过滤,滤液减压浓缩,得到浓缩液;所述超声的功率为200~400w,频率为30~50khz,时间为20~60min;
(b)采用甲醇水溶液将浓缩液进行定容,然后将定容液中的上清液进行微孔滤膜过滤,得到供试品溶液。
在本发明中,所述步骤(a)中药复方制剂与甲醇的用量比优选为1g:50~150ml,进一步优选为1g:70~130ml,更优选为1g:100ml;所述超声的功率优选为200~400w,进一步优选为220~380w,更优选为240~360w;所述超声的频率优选为30~50khz,更优选为35~45khz;所述超声的时间优选为20~60min,更优选为30~50min。在本发明中,所述步骤(a)中的过滤优选为常规滤纸过滤。本发明对减压浓缩的具体实施方式没有特别要求,采用本领域技术人员熟知的方式即可。
得到浓缩液后,本发明采用甲醇水溶液将浓缩液进行定容,然后将定容液中的上清液进行微孔滤膜过滤,得到供试品溶液。在本发明中,所述甲醇水溶液的体积浓度优选为10%~100%,进一步优选为30%~100%,更优选为50%~100%。在本发明的具体实施方式中,优选将浓缩液定容至5ml。在本发明中,所述微孔滤膜的孔径优选为0.20~0.50μm,进一步优选为0.25~0.45μm,更优选为0.3~0.45μm。
得到供试品溶液后,本发明采用高效液相色谱法对供试品溶液进行测试,根据《中药色谱指纹图谱相似度评价系统2012a版》,由得到的色谱数据生成中药复方制剂指纹图谱。
在本发明中,所述高效液相色谱法的色谱条件为:
色谱柱:c18色谱柱,优选为diamonsilc18色谱柱;
检测波长:200~220nm,优选为203nm;
流动相:流动相a为体积分数0.05%~0.3%的磷酸水溶液,优选为体积分数0.1%的磷酸水溶液;流动相b为乙腈;
洗脱方式为梯度洗脱,所述梯度洗脱的洗脱程序如表1所示;
所述色谱条件优选还包括:
流速:0.5~1.5ml/min,优选为1.0ml/min;
柱温:15~35℃,优选为25~30℃;
色谱柱规格:250mm×4.6mm,5μm;
进样体积:5~20μl,优选为10~15μl;
进样浓度:0.01~1.00g/ml,优选为0.10~0.50g/ml,更优选为0.10~0.30g/ml。
本发明采用与供试品溶液相同的高效液相色谱条件对混合对照品溶液进行测试,得到混合对照品溶液的色谱图;将混合对照品溶液的色谱图与由供试品得到的中药复方制剂指纹图谱进行对比,可指认中药复方制剂指纹图谱中的主要色谱峰。
在本发明中,所述混合对照品溶液的配制方法优选包括以下步骤:将人参皂苷re、人参皂苷rg1、喜树碱、人参皂苷rb1、表儿茶素和蟾蜍噻咛对照品溶于甲醇中,得到混合对照品溶液。
在本发明中,所述混合对照品溶液中,所述人参皂苷re的浓度优选为90~110μg/ml,所述人参皂苷rg1的浓度优选为60~80μg/ml,所述喜树碱的浓度优选为30~40μg/ml,所述人参皂苷rb1的浓度优选为280~320μg/ml,所述表儿茶素的浓度优选为35~45μg/ml,所述蟾蜍噻咛的浓度优选为15~25μg/ml。本发明优选将混合对照品溶液中各对照品的浓度控制在上述范围内,有利于使混合对照品溶液中各物质浓度与供试品溶液中对应物质浓度接近。
本发明还提供了上述技术方案所述方法得到的中药复方制剂指纹图谱,以人参皂苷rb1色谱峰为参照,所述中药复方制剂指纹图谱包括29个共有峰,具体如下:
1号峰,相对保留时间为0.064;
2号峰,相对保留时间为0.079;
3号峰,相对保留时间为0.088;
4号峰,相对保留时间为0.097;
5号峰,相对保留时间为0.108;
6号峰,相对保留时间为0.119;
7号峰,相对保留时间为0.134;
8号峰,相对保留时间为0.143;
9号峰,相对保留时间为0.160;
10号峰,相对保留时间为0.282;
11号峰,相对保留时间为0.294;
12号峰,相对保留时间为0.311;
13号峰,相对保留时间为0.341;
14号峰,相对保留时间为0.352;
15号峰,相对保留时间为0.411;
16号峰,相对保留时间为0.436;
17号峰,相对保留时间为0.520;
18号峰,相对保留时间为0.547;
19号峰,相对保留时间为0.570;
20号峰,相对保留时间为0.639;
21号峰,相对保留时间为0.667;
22号峰,相对保留时间为0.687;
23号峰,相对保留时间为0.898;
24号峰,相对保留时间为0.929;
25号峰,相对保留时间为0.939;
26号峰,相对保留时间为0.990;
27号峰,相对保留时间为1;
28号峰,相对保留时间为1.058;
29号峰,相对保留时间为1.169。
在本发明中,以人参皂苷rb1色谱峰为参照,所述中药复方制剂指纹图谱中共有峰的归属包括:
指纹图谱中的13号峰为蟾蜍噻咛;
17号峰为表儿茶素;
23号峰为人参皂苷re和rg1;
24号峰为喜树碱;
27号峰为人参皂苷rb1。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
本发明实施例中,使用的仪器、试剂如下:
仪器设备:agilent1260高效液相色谱仪,配置四元泵、dad检测器(美国agilent公司);jy5002电子天平(上海良平仪器仪表有限公司);xs205电子天平(瑞士mettler-toledo公司);re-52aa旋转蒸发器(上海亚荣生化仪器厂);shb-ⅲg循环水式多用真空泵(郑州长城科工贸有限公司);kq-300b型超声波清洗器(昆山市超声仪器有限公司)。
对照品、供试品及试剂:
对照品:人参皂苷re(批号:110754-201626,质量分数97.4%)、人参皂苷rg1(批号:110703-201731,质量分数93.6%)、喜树碱(批号:100532-201702,质量分数99.8%)、人参皂苷rb1(批号:110704-201827,质量分数91.2%)、表儿茶素(批号:110878-201703,质量分数99.7%),中国食品药品检定研究院;蟾蜍噻咛,自制。
供试品:本发明用中药复方制剂的批号分别为:20180516、20180517、20181205、20181218、20181224、20190118、20190121、20190122、20190123、20190124、20190125均为自制,上述批号供试品的制备方法相同,均为:
将干蟾皮、壁虎、金荞麦、喜树果、西洋参、仙鹤草和灵芝与甲醇混合后超声后过滤,滤液减压浓缩,得到浓缩液;所述干蟾皮、壁虎、金荞麦、喜树果、西洋参、仙鹤草和灵芝的质量比为1:1:8:6:2:4:3;所述超声的功率为300w,频率为40khz,时间为40min;采用甲醇水溶液将浓缩液进行定容,然后将定容液中的上清液进行微孔滤膜过滤,得到供试品溶液。
试剂:乙腈,色谱纯,美国tedia公司;水为纯水;其余试剂均为分析纯。
实施例1
精密度实验
取中药复方制剂(20190122批),精密称取中药复方制剂粉末0.5g,加入50ml甲醇作为提取溶剂,在功率300w,频率40khz下超声处理40min,冷却,摇匀,过滤,滤液减压浓缩,用体积浓度50%的甲醇水溶液溶解并定容至5ml,吸取上清液,使其过0.45μm的微孔滤膜,得供试品溶液。
采用下列高效液相色谱条件进行测试:
色谱柱:diamonsilc18色谱柱;
检测波长:203nm;
流动相:流动相a为体积分数0.1%的磷酸水溶液;流动相b为乙腈;
洗脱方式为梯度洗脱,所述梯度洗脱的洗脱程序如表1所示;
流速:1.0ml/min;
柱温:30℃;
色谱柱规格:250mm×4.6mm,5μm;
进样体积:10μl;
进样浓度:0.10g/ml。
按照上述色谱条件连续进样6针,以人参皂苷rb1峰为参照峰,计算各共有峰与参照峰的相对峰面积及相对保留时间,并计算rsd值,结果如表2和表3所示:
表2精密度实验(各共有峰的相对峰面积)
注:(s)为参照峰
表3精密度实验(各共有峰的相对保留时间)
由表2和表3可知,各共有峰的相对峰面积rsd%<5%、相对保留时间rsd%<5%,说明本发明提供的检测方法精密度良好。
实施例2
重复性实验
取中药复方制剂(20190122批),平行称取6份,按实施例1的供试品溶液的制备方法制备6份供试品溶液,按实施例1中色谱条件分别进样,以人参皂苷rb1峰为参照峰,计算各共有峰与参照峰的相对峰面积及相对保留时间,并计算rsd值,结果如表4和表5所示:
表4重复性实验(各共有峰的相对峰面积)
表5重复性实验(各共有峰的相对保留时间)
由表4和表5可知,各共有峰的相对峰面积rsd%<5%、相对保留时间rsd%<5%,说明该方法重复性良好。
实施例3
稳定性实验
取中药复方制剂(20190122批),按实施例1的供试品溶液的制备方法制备供试品溶液,按实施例1中色谱条件,分别于0h、3h、6h、9h、12h、24h条件进样,以人参皂苷rb1峰为参照峰,计算各共有峰与参照峰的相对峰面积及相对保留时间,并计算rsd值,结果如表6和表7所示:
表6稳定性实验(各共有峰的相对峰面积)
表7稳定性实验(各共有峰的相对保留时间)
由表6和表7可知,各共有峰的相对峰面积rsd%<5%、相对保留时间rsd%<5%,说明供试品溶液在24h内基本稳定。
实施例4
中药复方制剂指纹图谱的建立
(1)供试品溶液的制备
取以上11个批次的中药复方制剂,批号依次为20180516、20180517、20181205、20181218、20181224、20190118、20190121、20190122、20190123、20190124、20190125,分别编号为s1、s2、s3、s4、s5、s6、s7、s8、s9、s10、s11,分别精密称取粉末0.5g,加入50ml甲醇作为提取溶剂,在功率300w,频率40khz下超声处理40min,冷却,摇匀,过滤,滤液减压浓缩,用体积浓度50%甲醇溶解并定容至5ml,吸取上清液,使其过0.45μm的微孔滤膜,得11个批次的中药复方制剂供试品溶液。
(2)混合对照品溶液的制备
取人参皂苷re、人参皂苷rg1、喜树碱、人参皂苷rb1、表儿茶素、蟾蜍噻咛对照品,精密称定,置于10ml容量瓶中,用甲醇溶解并定容到刻度,震荡摇匀,得到混合对照品溶液。其中混合对照品溶液中人参皂苷re、人参皂苷rg1、喜树碱、人参皂苷rb1、表儿茶素和蟾蜍噻咛的质量浓度依次为99.54μg/ml、71.88μg/ml、33.88μg/ml、306.38μg/ml、41.83μg/ml和19.60μg/ml。
(3)高效液相色谱条件
色谱柱:diamonsilc18柱(规格为250mm×4.6mm,5μm);
流动相:流动相a为体积分数0.1%的磷酸水溶液,流动相b为乙腈;
洗脱方式为梯度洗脱,梯度洗脱方式如表1所示;
流速:1.0ml/min;
柱温:30℃;
进样量:10μl;
检测波长:203nm。
(4)分别精密吸取供试品溶液和对照品溶液各10μl,采用上述高效液相色谱条件进行测试,分别得到供试品溶液的液相色谱和对照品溶液的液相色谱。
对照品溶液的液相色谱如图1所示,图1中1号峰为蟾蜍噻咛对照品,2号峰为表儿茶素对照品,3号峰为人参皂苷re和人参皂苷rg1对照品,4号峰为喜树碱对照品,5号峰为人参皂苷rb1对照品。
将供试品溶液的液相色谱导入指纹图谱相似度评价软件《中药色谱指纹图谱相似度评价系统2012a版》分析,以s8供试品作为对照图谱,选取“时间窗宽度”为0.5min,采用平均数法计算,多点校正、数据匹配,生成叠加色谱图,即中药复方制剂指纹图谱,如图2所示;以11批供试品图谱为基础的“共有模式”生成的对照图谱如图3所示。将11批制剂的指纹图谱与生成的对照图谱进行相似度分析,计算各共有峰与参照峰的相对保留时间,并计算rsd值。结果如表8、表9所示:
表811批制中药复方剂指纹图谱相似度
表911批次中药复方制剂指纹图谱相对保留时间测定结果
由表8和表9可知,不同批次中药复方制剂指纹图谱与对照图谱的相似度均在0.9之上,表明不同批次间的相似度较高;不同批次中药复方制剂各共有峰相对保留时间rsd%<5%,表明本发明提供的测试方法稳定性较好。
实施例5
按实施例1方法分别测定中药复方制剂中壁虎、喜树果、西洋参、干蟾皮、灵芝、仙鹤草、金荞麦的阳性对照色谱图及阴性对照色谱图,通过dad检测器对中药复方制剂指纹图谱与各药材阳性对照色谱图及阴性对照色谱图中色谱峰的紫外吸收进行分析,比较色谱峰的保留时间,最终确认中药复方制剂指纹图谱中共有峰的归属峰。结果如表10所示:
表10共有色谱峰在药材图谱上的归属峰
(一)壁虎中的共有峰归属分析
壁虎药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图如图4所示;壁虎药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图如图4所示。图4中i为壁虎阴性供试品,j为壁虎药材供试品,o为实施例4得到的中药复方制剂指纹图谱。通过色谱图对比,可知中药复方制剂指纹图谱共有峰中的7号峰来自壁虎药材。
(二)喜树果中的共有峰归属分析
喜树果药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图如图4所示;喜树果药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图如图4所示。图4中c为喜树果阴性供试品,d为喜树果药材供试品。通过色谱图对比,可知中药复方制剂指纹图谱共有峰中的11号、15号、24号、26号峰来自喜树果药材。
(三)西洋参中的共有峰归属分析
西洋参药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图如图4所示;西洋参药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图如图4所示。图4中a为西洋参阴性供试品,b为西洋参药材供试品。通过色谱图对比,可知中药复方制剂指纹图谱共有峰中的23号、27号峰来自西洋参药材。
(四)金荞麦中的共有峰归属分析
金荞麦药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图如图4所示;金荞麦药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图如图4所示。图4中e为金荞麦阴性供试品,f为金荞麦药材供试品。通过色谱图对比,可知指纹图谱共有峰中的14号峰来自金荞麦药材。
(五)仙鹤草中的共有峰归属分析
仙鹤草药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图如图4所示;仙鹤草药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图如图4所示。图4中m为仙鹤草阴性供试品,n为仙鹤草药材供试品。通过色谱图对比,可知指纹图谱共有峰中的18号、19号、20号、22号、25号峰来自仙鹤草药材。
(六)灵芝中的共有峰归属分析
灵芝药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图如图4所示;灵芝药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图如图4所示。图4中k为灵芝阴性供试品,l为灵芝药材供试品。通过色谱图对比,可知指纹图谱共有峰中的28号峰来自灵芝药材。
(七)干蟾皮中的共有峰归属分析
干蟾皮药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图如图4所示;干蟾皮药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图如图4所示。图4中g为干蟾皮阴性供试品,h为干蟾皮药材供试品。通过色谱图对比,可知指纹图谱共有峰中的13号峰来自干蟾皮药材。
图4中峰的归属为:13号峰对应蟾蜍噻咛对照品,17号峰对应表儿茶素对照品,23号峰对应人参皂苷re和人参皂苷rg1对照品,24号峰对应喜树碱对照品,27号峰对应人参皂苷rb1对照品。
对比例1
按照实施例4中s1批次供试品溶液的配制方法以及测试方法进行实验,区别在于,对比例1分别在检测波长225nm、254nm、280nm、296nm条件下进行实验。将实施例4中s1批次供试品溶液的色谱图与对比例1中不同检测波长下供试品的色谱图进行对比,结果如图5所示。图5中a为实施例4检测波长203nm的色谱图,b对比例1检测波长为225nm的色谱图,c为对比例1检测波长为254nm的色谱图,d为对比例1检测波长为280nm的色谱图,e对比例1检测波长为296nm的色谱图。由图5可知,本发明将检测波长设置为203nm,能够检测的色谱峰数量最多、色谱峰响应值较高、基线较平稳。
对比例2
按照实施例4中s1批次供试品溶液的配制方法以及测试方法进行实验,测试方法中梯度洗脱程序与实施例4不同,对比例2的梯度洗脱程序见表11;对比例2分别在流动相a为水、流动相a为体积分数0.2%的磷酸水溶液、流动相a为体积分数0.3%的磷酸水溶液条件下进行实验。将实施例4中s1批次供试品溶液的色谱图与对比例2中不同流动相a下供试品的色谱图进行对比,结果如图6所示。图6中a为对比例2流动相a为水的色谱图,b为实施例4流动相a为体积分数0.1%磷酸水溶液的色谱图,c为对比例2流动相a为体积分数0.2%磷酸水溶液的色谱图,d为对比例2流动相a为体积分数0.3%磷酸水溶液的色谱图。由图6可知,本发明采用表1所示梯度洗脱程序,并将流动相a设置成体积分数0.1%的磷酸水溶液,能够检测的色谱峰数量最多、色谱峰响应值较高、基线较平稳。
表11梯度洗脱程序
对比例3
按照实施例4中s1批次供试品溶液的配制方法以及测试方法进行实验,测试方法中梯度洗脱程序与实施例4不同,对比例3的梯度洗脱程序见表12;对比例3分别在水作为提取溶剂、体积分数为50%的甲醇作为提取溶剂的条件下进行实验。将实施例4中s1批次供试品溶液的色谱图与对比例3中不同提取溶剂下供试品的色谱图进行对比,结果如图7所示。图7中a为对比例3提取溶剂为水的色谱图,b为对比例3提取溶剂为体积分数50%甲醇水溶液的色谱图,c为实施例4提取溶剂为甲醇的色谱图。由图7可知,本发明采用表1所示梯度洗脱程序,并采用甲醇作为提取溶剂,能够检测的色谱峰数量最多、色谱峰响应值较高、基线较平稳。
表12梯度洗脱程序
综上,本发明提供的中药复方制剂指纹图谱中的29个共有峰,在干蟾皮、壁虎、喜树果、西洋参、灵芝、仙鹤草、金荞麦7味药材阳性对照与阴性对照色谱图中,基本找到明确归属。由此可见,制剂的指纹图谱基本能代表制剂的物质基础。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!