葡糖鞘氨醇的检测方法与流程
本发明涉及分析检测
技术领域:
,特别是涉及一种葡糖鞘氨醇的检测方法。
背景技术:
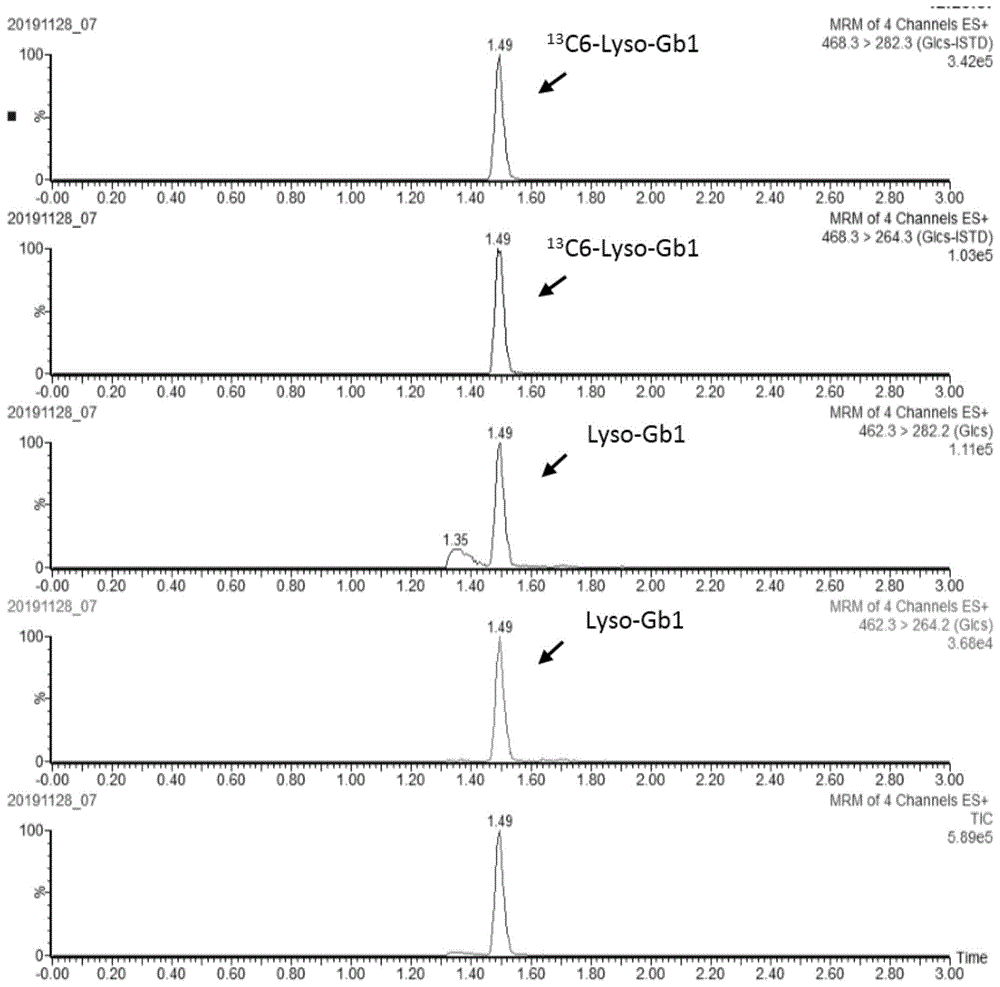
:戈谢病(gaucherdisease,gd)是一种常染色体隐性溶酶体疾病,是由gba1基因突变导致的β-葡萄糖脑苷酶活性受损引起的。葡糖鞘氨醇(glucosylsphingosine,lyso-gb1)是一种脱酰形式的葡萄糖神经酰胺,已被公认为诊断和监测戈谢病患者的高度敏感和特异性的生物标志物。目前,现有的葡糖鞘氨醇的检测方法中,检测样本多基于血浆或尿液,色谱条件较复杂,且对仪器要求较高。技术实现要素:基于此,有必要针对现有的葡糖鞘氨醇的检测方法中,检测样本多基于血浆或尿液,色谱条件较复杂,且对仪器要求较高的问题,提供一种葡糖鞘氨醇的检测方法。一种葡糖鞘氨醇的检测方法,包括如下步骤:获取干血斑样本;向所述干血斑样本中加入含有葡糖鞘氨醇同位素内标的乙腈水溶液,振荡即得待测样品;采用液相色谱串联质谱法检测所述待测样品。在其中一个实施例中,所述乙腈水溶液中乙腈与水的体积比为50:(45~55)。在其中一个实施例中,所述液相色谱串联质谱法的液相色谱条件包括:色谱柱:c18柱,规格为2.1×50mm,1.7μm;流动相a:体积浓度为0.09%~0.11%的甲酸水溶液,流动相b:体积浓度为0.09%~0.11%的甲酸乙腈溶液;采用梯度洗脱。在其中一个实施例中,所述梯度洗脱包括如下程序:0~2min:流动相b的体积百分比由45%上升至65%,流动相a的体积百分比由55%降低至35%;2min~2.1min:流动相b的体积百分比由65%b上升至100%,流动相a的体积百分比由35%降低至0;2.1min~4min:流动相b的体积百分比保持100%不变,流动相a的体积百分比保持0不变;4min~4.1min:流动相b的体积百分比由100%降低至45%,流动相a的体积百分比由0上升至55%;4.1min~5min:流动相b的体积百分比保持45%不变,流动相a的体积百分比保持55%不变。在其中一个实施例中,所述液相色谱串联质谱法的液相色谱条件还包括:流速:0.3ml/min~0.5ml/min;进样量:5μl~10μl;柱温:40℃~50℃。在其中一个实施例中,所述液相色谱串联质谱法的质谱条件包括:离子源:电喷雾离子源,正离子模式;毛细管电压:0.6kv~1.0kv;锥孔电压:30v~40v;碰撞电压:15v~30v;离子源温度:145℃~155℃;脱溶剂气温度:550℃~650℃;脱溶剂气流量:800l/hr~1000l/hr;锥孔气流量:45l/hr~55l/hr;离子扫描模式:多重反应监测模式。在其中一个实施例中,所述乙腈水溶液中的葡糖鞘氨醇同位素内标为13c6-葡糖鞘氨醇。在其中一个实施例中,所述液相色谱串联质谱法的质谱条件还包括:葡糖鞘氨醇的检测离子通道为462.3>264.2和462.3>282.2,其中462.3>264.2为定量离子通道,462.3>282.2为定性离子通道;13c6-葡糖鞘氨醇的检测离子通道为468.3>264.3和468.3>282.3,其中468.3>264.3为定量离子通道,468.3>282.3为定性离子通道。在其中一个实施例中,所述干血斑样本的直径为3mm~3.5mm;所述乙腈水溶液的加入量为80μl~100μl。在其中一个实施例中,所述振荡的时间为20min~50min,速度为600rpm~700rpm本发明的葡糖鞘氨醇的检测方法,基于液相色谱串联质谱技术,以干血斑作为检测样本,采用葡糖鞘氨醇同位素内标作为内标物,并通过乙腈水溶液振荡提取,其前处理简单,所需样本量少,易采集、运输及储存。进一步地,该检测方法采用c18柱色谱柱,其色谱流动相较简单。且通过验证,该检测方法简单灵敏、快速准确、重复性好、精密度、基质效应及加标回收率满足检测要求,可用于戈谢病的快速诊断及监测。附图说明图1为实施例1检测的5ng/ml的标准品溶液(含2ng/ml内标溶液)在不同离子通道检测的色谱图;图2为实施例1检测的7个标准品溶液检测的色谱图;图3为实施例1检测的干血斑样本在不同离子通道检测的色谱图;图4为实施例1检测的葡糖鞘氨醇的标准曲线图;图5为实施例1检测的干血斑样本的线性回归曲线;图6为实施例2检测的1ng/ml的标准品溶液(含1ng/ml内标溶液)在不同离子通道检测的色谱图;图7为实施例3检测的1ng/ml的标准品溶液(含1ng/ml内标溶液)在不同离子通道检测的色谱图;图8为实施例4检测的1ng/ml的标准品溶液(含1ng/ml内标溶液)在不同离子通道检测的色谱图。具体实施方式为使本发明的上述目的、特征和优点能够更加明显易懂,对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施例的限制。除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的
技术领域:
的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“及/或”包括一个或多个相关的所列项目的任意的和所有的组合。本发明提供一种葡糖鞘氨醇的检测方法,包括如下步骤:获取干血斑样本;向干血斑样本中加入含有葡糖鞘氨醇同位素内标的乙腈水溶液,振荡即得待测样品;采用液相色谱串联质谱法检测待测样品。在本发明中,干血斑样本可通过常规的方法进行制作得到。例如:新生儿生后3~7天在知情同意下采集足跟血,滴至whatman903滤纸片上自由扩散形成血斑,自然晾干,充分干燥(约4小时),制成干血斑,放入密封袋中,置于-20℃~4℃冰箱避光保存待用。在一些实施例中,采用96孔v型截底透明微孔板对干血斑样本进行前处理。前处理为:向干血斑样本中加入含有葡糖鞘氨醇同位素内标的乙腈水溶液,振荡。通过使用96孔v型截底透明微孔板进行前处理可明显减少样本制备的时间,同时可减少移液、装卸吸嘴等操作,减少耗材的使用,简化操作步骤,降低移液错误风险,节约样本制备的时间。同时还能更好的保护仪器,明显降低仪器的维护成本。在一些实施例中,乙腈水溶液中乙腈与水的体积比为50:(45~55)。优选地,乙腈水溶液中乙腈与水的体积比为50:50。在一些实施例中,干血斑样本的直径为3mm~3.5mm;乙腈水溶液的加入量为80μl~100μl。振荡提取的时间为20min~50min,速度为600rpm~700rpm。进一步地,采用pepanthera-pμnchertm9打孔器打一个干血斑(3.2mm)样本,置于96孔v型截底透明微孔板中,加入100μl含有葡糖鞘氨醇同位素内标的乙腈水溶液,用粘性塑料封套覆盖微孔板,确保密封良好,将挥发量降低到最小,置于振荡器,在45℃条件下振荡20分钟,微孔板振荡速度为650rpm,然后取85μl的待测样品转移到v型底耐热微孔板内,用铝箔封套覆盖微孔板,尽量密封,待进样分析。在一些实施例中,液相色谱串联质谱法的液相色谱条件包括:色谱柱:c18柱,规格为2.1×50mm,1.7μm;流动相a:体积浓度为0.09%~0.11%的甲酸水溶液,流动相b:体积浓度为0.09%~0.11%的甲酸乙腈溶液;采用梯度洗脱。优选地,流动相a:体积浓度为0.1%的甲酸水溶液,流动相b:体积浓度为0.1%的甲酸乙腈溶液。进一步地,梯度洗脱包括如下程序:0~2min:流动相b的体积百分比由45%上升至65%,流动相a的体积百分比由55%降低至35%;2min~2.1min:流动相b的体积百分比由65%b上升至100%,流动相a的体积百分比由35%降低至0;2.1min~4min:流动相b的体积百分比保持100%不变,流动相a的体积百分比保持0不变;4min~4.1min:流动相b的体积百分比由100%降低至45%,流动相a的体积百分比由0上升至55%;4.1min~5min:流动相b的体积百分比保持45%不变,流动相a的体积百分比保持55%不变。需要说明的是,进样后1.3min~1.8min流动相通过切换阀切换进入质谱检测,其余时间切换至废液。在一些实施例中,液相色谱串联质谱法的液相色谱条件还包括:流速:0.3ml/min~0.5ml/min;进样量:5μl~10μl;柱温:40℃~50℃。优选地,流速:0.4ml/min;进样量:10μl;柱温:45℃。在一些实施例中,液相色谱串联质谱法的质谱条件包括:离子源:电喷雾离子源,正离子模式;毛细管电压:0.6kv~1.0kv;锥孔电压:30v~40v;碰撞电压:15v~30v;离子源温度:145℃~155℃;脱溶剂气温度:550℃~650℃;脱溶剂气流量:800l/hr~1000l/hr;锥孔气流量:45l/hr~55l/hr;离子扫描模式:多重反应监测模式。优选地,毛细管电压:0.8kv;锥孔电压:35v;碰撞电压:20v;离子源温度:150℃;脱溶剂气温度:600℃;脱溶剂气流量:1000l/hr;锥孔气流量:50l/hr。在一些实施例中,乙腈水溶液中的葡糖鞘氨醇同位素内标为13c6-葡糖鞘氨醇。在一些实施例中,液相色谱串联质谱法的质谱条件还包括:葡糖鞘氨醇的检测离子通道为462.3>264.2和462.3>282.2,其中462.3>264.2为定量离子通道,462.3>282.2为定性离子通道;13c6-葡糖鞘氨醇的检测离子通道为468.3>264.3和468.3>282.3,其中468.3>264.3为定量离子通道,468.3>282.3为定性离子通道。以下为具体实施例实施例1一、溶液的制备1、标准品溶液:精密称取葡糖鞘氨醇标准品,用乙腈溶解,制成浓度为50μg/ml的标准品储备液,置于-20℃的冰箱中保存。使用时再用乙腈稀释制成0.4、2、5、10、50、250、500ng/ml的标准系溶液。2、含内标物的萃取液:精密称取13c6-葡糖鞘氨醇,用乙腈溶解并稀释至1μg/ml,接着用体积浓度为50%的乙腈水溶液稀释成含2ng/ml13c6-葡糖鞘氨醇的萃取液。二、干血斑质控品制备1、取标准品储备液稀释成浓度为250ng/ml、10μg/ml、50μg/ml的标准品质控溶液。2、采集健康志愿者的静脉血2ml,edta抗凝,采集后直接将0ng/ml、250ng/ml、10μg/ml、50μg/ml的标准品质控溶液按1:9的体积比分别添加到全血中混匀,得到空白、低、中、高浓度的全血质控溶液。分别取空白、低、中、高浓度的全血质控溶液75μl滴至干血斑,自由扩散形成血斑,自然晾干,待血斑充分干燥后(约4小时),得空白、低、中、高浓度的干血斑质控品,将干血斑质控品放入密封袋中,置于4℃~-20℃冰箱避光保存待用。三、干血斑样本的处理方法将干血斑质控品、干血斑样本和标准品溶液采用如下方法处理:取干血斑质控品、干血斑样本,用pepanthera-pμnchertm9打孔器打一个血斑(3.2mm)分别置于96孔v型截底透明微孔板中,留7个孔不打血斑,然后均分别加入100μl前述含内标物的萃取液;未打血斑的7个孔中再分别加入10μl已配制好的浓度为0.4ng/ml、2ng/ml、5ng/ml、10ng/ml、50ng/ml、250ng/ml、500ng/ml的标准品溶液,用粘性塑料封套覆盖微孔板,确保密封良好,将挥发量降低到最小。置于振荡器,在45℃条件下振荡20分钟,微孔板振荡速度为650rpm。然后取85μl的各溶液转移到v型底耐热微孔板内,用铝箔封套覆盖微孔板,尽量密封,进样分析。四、液相色谱串联质谱检测条件仪器:uplci-classacquityxevotqd超高效液相色谱串联质谱联用仪(waters)1、色谱条件:色谱柱:acquityuplc@behc181.7μm,2.1×50mm;流动相a:体积浓度为0.1%甲酸水溶液,流动相b:体积浓度为0.1%甲酸乙腈溶液;柱温45℃;流速0.4ml/min;进样量10μl;检测时间5min;采用梯度洗脱方式;梯度洗脱的程序:0~2min:流动相b的体积百分比由45%上升至65%,流动相a的体积百分比由55%降低至35%;2min~2.1min:流动相b的体积百分比由65%b上升至100%,流动相a的体积百分比由35%降低至0;2.1min~4min:流动相b的体积百分比保持100%不变,流动相a的体积百分比保持0不变;4min~4.1min:流动相b的体积百分比由100%降低至45%,流动相a的体积百分比由0上升至55%;4.1min~5min:流动相b的体积百分比保持45%不变,流动相a的体积百分比保持55%不变;进样后1.3~1.8min流动相通过切换阀切换进入质谱检测,其余时间切换至废液。2、质谱条件:离子源:电喷雾离子源(esi),正离子模式;毛细管电压:0.8kv;锥孔电压35v;离子源温度:150℃,脱溶剂气温度:600℃,脱溶剂气流速:1000l/hr;锥孔气:50l/hr;离子扫描模式:多重反应监测(mrm)模式,具体如下表1:表1化合物离子通道锥孔电压(v)碰撞电压(v)葡糖鞘氨醇(lyso-gb1)462.3>264.23520葡糖鞘氨醇(lyso-gb1)462.3>282.2352013c6-葡糖鞘氨醇(13c6-lyso-gb1)468.3>264.3352013c6-葡糖鞘氨醇(13c6-lyso-gb1)468.3>282.33520五、数据分析在上述液相色谱串联质谱检测条件下,一低浓度(5ng/ml)标准品(含2ng/ml内标溶液)的不同离子通道检测色谱图如图1所示;上述7个标准品溶液(含2ng/ml内标溶液)检测的色谱图如图2所示;一患者干血斑样本的不同离子通道的色谱图如图3所示;其中,葡糖鞘氨醇的保留时间rt为1.49min。采用同位素内标法进行定量分析,利用分析软件以7个标准品与其内标物的浓度比为x轴,标准品与内标物峰面积比为y轴,建立标准曲线,由于标准曲线未制作成干血斑标曲,根据体积换算,则干血斑待测物的浓度在建立的标准曲线基础上乘以10/3.3计算得出,标准曲线如图4所示,y=0.0718x±0.0017,r=0.997,r2=0.994。方法验证:1、精密度实验选取低(2.5ng/ml)、中(100ng/ml)、高浓度(500ng/ml)的干血斑质控品(记为lc、mc、hc),按照前述干血斑质控品的处理方法进行处理,每个干血斑质控品一天内连续测定六次,根据质控品中葡糖鞘氨醇与其内标峰的面积比,代入前述标准曲线,得到质控品中葡糖鞘氨醇的浓度,计算相对标准偏差,即批内精密度(cv)介于2%-9%之间(见表2-1和2-2);连续测定5天,计算相对标准偏差,即批间精密度(cv)介于3%-7%之间(见表3)。表2-1表2-2表3天数lcmchc13.83816797.047569.340523.97033396.20733559.626333.307590.25383531.577843.82199.91617592.348354.00283395.7625550.2773average3.78796795.83737560.6341sd0.2801273.51490222.55071cv(%)7.3951843.667574.022359重复性实验:选取低(5ng/ml)、中(10ng/ml)、高浓度(500ng/ml)的干血斑质控品(记为qc1、qc2、qc3),按照前述干血斑质控品的处理方法进行处理,每个质控品连续测定不少于20次,根据质控品中葡糖鞘氨醇与其内标峰的面积比,代入前述标准曲线,得到质控品中葡糖鞘氨醇的浓度,计算相对标准偏差介于1%-5%之间,见表4。表4次数qc1qc2qc317.77213.272484.22528.07913.492474.61238.1713.355492.99248.86413.988488.07658.44714.195496.42768.93714.734468.8178.80113.694490.41688.87414.184493.51398.78414.024472.651107.95713.418497.717118.70113.898483.158128.55514.017476.675138.61614.044498.512148.78414.134504.442158.82413.444489.302168.56513.69490.152178.68713.815482.336188.98414.359484.46198.81314.261496.71208.11814.532491.567average8.5713.93487.84sd0.360.409.45cv(%)4.172.901.94准确度实验:选取低(2.5ng/ml)、中(100ng/ml)、高浓度(500ng/ml)的干血斑质控品(记为lc、mc、hc),按照前述干血斑质控品的处理方法进行处理,每个浓度质控品各5份分别进行加标(50ppb标准品)回收实验,根据质控品中葡糖鞘氨醇与其内标峰的面积比,代入前述标准曲线,得到质控品中葡糖鞘氨醇的浓度,通过每个浓度的5份质控品结果计算平均回收率,其平均回收率介于102%-113%之间,其偏差率在±15%之间,见表5,回收率的计算公式为:回收率(%)=检测浓度/理论浓度×100%。表5序号lc+50ppbmc+50ppbhc+50ppb1159.884274.8727.0422172.01301.539644.6153168.085266.991719.6844169.166271.439771.2585163.319270.808693.664ave166.4928277.1154711.2526理论浓度153.81245.4086693.2664回收率(%)108113102偏差率(%)8.2512.922.59基质效应:选取一份病人干血斑样本(记为m)、一份质控品(记为qc1)及纯溶剂各5份,m和qc1分别按照前述干血斑样本和干血斑质控品的处理方法处理,纯溶剂按照前述标准品溶液的处理方法处理,然后分别加入50ppb的标准品,根据样本中葡糖鞘氨醇与其内标峰的面积比,代入前述标准曲线,得到样本中葡糖鞘氨醇的浓度,其中,纯溶剂为体积浓度为50%的乙腈水溶液。比较天然基质与纯溶剂的信号比值,其比值分别为0.92和0.94,基质效应cv分别为3.75%和9.31%。线性:选择7个浓度的病人干血斑样本,按照前述干血斑样本的处理方法进行处理,根据葡糖鞘氨醇与其内标峰的面积比,代入前述标准曲线,得到实测葡糖鞘氨醇的浓度,每个样本重复测定2次,见表6,使用多元回归方程评价线性,线性方程为y=1.0616x-9.1971,r2=0.9984,如图5所示。表6从上述检测结果可知,本发明的葡糖鞘氨醇检测方法简单灵敏、快速准确、重复性好、精密度、基质效应及加标回收率满足检测要求。实施例2与实施例1基本相同,区别在于,流动相a:10mm甲酸铵水溶液、体积浓度为90%的乙腈水溶液和体积浓度为0.01%甲酸,流动相b:10mm甲酸铵水溶液和体积浓度为0.01%甲酸;毛细管电压:3.5kv。如图6所示,检测的1ng/ml的标准品溶液(含1ng/ml内标溶液)的总离子流图(tic)信号强度为8.53e4。实施例3与实施例2基本相同,区别在于,流动相a:体积浓度为0.1%的甲酸水溶液,流动相b:甲醇。如图7所示,检测的1ng/ml的标准品溶液(含1ng/ml内标溶液)的总离子流图(tic)信号强度9.58e4。实施例4与实施例1基本相同,区别在于,毛细管电压:3.5kv,梯度洗脱程序为:0~2min:流动相b的体积百分比由50%上升至65%,流动相a的体积百分比由50%降低至35%。如图8所示,检测的1ng/ml标准品溶液(含1ng/ml内标溶液)的总离子流图(tic)信号强度1.30e5。以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1