一种基于深度学习模型的尿液蛋白质组学谱图数据分析系统的制作方法
本发明涉及蛋白质组学分析领域,特别是涉及一种基于深度学习模型的尿液蛋白质组学谱图数据分析系。
背景技术:
尿液是血液经肾小球滤过,经肾小管和集合管重吸收、排泻及分泌产生的终末代谢产物,其组成与性状可反映整个机体的状况,是临床最常用的检测样本之一。尿液中蛋白质种类和数量的变化携带有某种疾病发生、发展及预后的信息,可作为研究尿液生物标志物的理想来源,也可以在一定程度上反映血液和整个机体的状态。此外,与血液样本相比,尿液样本具有获取无创、可大量获取、不受稳态调节、可以容纳并积累更多变化的特点。因此,尿液蛋白质组学作为诠释尿液蛋白所携带信息的最有效方法,获得了广泛的关注。
目前,常用于尿液蛋白质组学分离鉴定的方法主要有二维聚丙烯酰胺凝胶电泳-质谱联用(2dpage-ms)、液相色谱-质谱联用(lc-ms)、蛋白质芯片-质谱联用(pc-ms)和毛细管电泳-质谱联用(ce-ms)等。其中,液相色谱-串联质谱技术(简称lc-ms/ms)作为一种联用技术,结合了液相色谱的高效分离能力和质谱采集的待检测分子的精确分子量及强度信息。该技术具有灵敏度高、选择性强的优势,可以获得丰富的蛋白质定性和定量信息,同时方便建立快速、高效的蛋白质组检测体系,因而在尿液蛋白质组学分析领域具有显著的优势。
但是,由于不同个体的尿液样品中蛋白质丰度跨度宽、个体差异性大,导致不同个体的尿液蛋白质组学分析缺乏可比性,极大的限制了尿液蛋白质组学分析方法在尿液生物标志物的检测及相关疾病的辅助诊断中的应用。
技术实现要素:
针对上述问题,本发明的目的在于提供一种能够对尿液蛋白质组的谱图数据解析和识别,从而高准确度预测该尿液样本对应的对象是健康人还是某种疾病患者的分析系统,以及该分析系统在开发尿液蛋白质异常的相关疾病的辅助诊断工具中的用途。
本发明提供了一种基于深度学习模型的蛋白质组学谱图数据分析系统,所述系统包括以下三个部分:
第一部分:一致性处理部分,在该部分将样本的蛋白质组学谱图数据进行一致性处理,得到标准化后的峰度矩阵;一致性处理的方法包括以下步骤:
(a)将样本的蛋白质组学谱图数据编号,依次记为1,2,……,n-1,n;n为大于1的整数;然后根据样本的来源,分别对蛋白质组学谱图数据进行分类标记,将来源于健康人和患者样本的蛋白质组学谱图数据区分开;
(b)将步骤(a)中的每个蛋白质组学谱图数据按照横坐标均分成x份,记为x个窗口,将每个窗口中的所有峰值数据叠加,记为峰值数据之和,得到具有x个窗口、且每个窗口只有一个峰值数据之和的峰度矩阵;其中,x为大于等于1的整数;
(c)在步骤(b)所得的峰度矩阵中,将每个蛋白质组学谱图数据中每个窗口的峰值数据之和除以该谱图数据中最大的峰值,记为标准化后的峰值数据之和,得到标准化后的峰度矩阵;
第二部分:深度学习模型构建部分;
第三部分:深度学习模型的训练和预测部分。
进一步地,步骤(a)中,所述样本为尿液样本,优选为人的尿液样本;
和/或,所述蛋白质组学谱图数据为蛋白质组学液相色谱-质谱联用技术检测数据,所述液相色谱-质谱联用技术检测数据优选为原始液相色谱-串联质谱技术检测数据,更优选为一级原始液相色谱-串联质谱技术检测数据;
和/或,一个样本的蛋白质组学谱图数据为一个或多个。
进一步地,步骤(a)中,所述分类标记的方法为:将来源于健康人的尿液样本的蛋白质组学谱图数据标记为q,将来源于患者的尿液样本的蛋白质组学谱图数据标记为p,q与p不同;
优选的,所述来源于患者的尿液样本的蛋白质组学谱图数据中,将来源于轻度患者的尿液样本的蛋白质组学谱图数据标记为p1,和/或,将来源于中度患者的尿液样本的蛋白质组学谱图数据标记为p2,和/或,将来源于重度患者的尿液样本的蛋白质组学谱图数据标记为p3,p1、p2、p3、p互不相同。
进一步地,步骤(b)中,所述横坐标为质荷比;x为大于等于100的整数,优选为大于等于700小于等于800的整数;
和/或,步骤(b)中,所述峰度矩阵的计算公式如式(1)所示:
其中,im为峰度矩阵;i为蛋白质组学谱图数据的序号,1≤i≤n;j为窗口的序号,1≤j≤x;k为第i个蛋白质组学谱图数据、第j个窗口中峰的序号,
和/或,步骤(c)中,所述标准化后的峰度矩阵的计算公式如式(2)所示:
nim=im/(max(i1,1…j),…,max(ii,1…j))(2)
其中,nim为标准化后的峰度矩阵,max(ii,1…j)为序号为i的蛋白质组学谱图数据中最大的峰值,im表示步骤(b)计算得到的峰度矩阵。
进一步地,第二部分中,所述深度学习模型为深度神经网络模型,优选的,所述深度神经网络模型是基于keras框架搭建的,更优选的,所述深度神经网络模型内嵌了一个初级的深度神经网络模型。
进一步地,所述初级的深度神经网络模型包括以下组成部分:输入层、隐藏层和输出层;优选的,所述隐藏层为3层,第一层有128个节点、第二层有64个节点、第三层有32个节点;更优选的,所述隐藏层中激活函数为线性整流函数relu,输出层激活函数为归一化指数函数softmax。
进一步地,第三部分中,所述深度学习模型的训练和预测的方法包括以下步骤:将第一部分得到的标准化的峰度矩阵分为训练集和测试集,先利用训练集中的标准化后的峰度矩阵对第二部分构建的深度学习模型进行训练,再利用训练后的深度学习模型对测试集中的标准化后的峰度矩阵进行预测,预测测试集对应的样本的来源。
进一步地,所述训练集中的数据个数为n1,测试集中的数据个数为n-n1,n-n1为大于等于1且小于n的整数,优选为1。
本发明还提供的上述基于深度学习模型的蛋白质组学谱图数据分析系统在制备疾病辅助诊断工具中的用途。
进一步地,所述疾病为与尿蛋白质异常相关的疾病,优选为受新型冠状病毒sars-cov-2感染的疾病,更优选为受新型冠状病毒sars-cov-2感染的呼吸道疾病。
本发明中,原始液相色谱-质谱联用技术检测数据是指未经处理的液相色谱-质谱联用技术检测的谱图数据,一级原始液相色谱-串联质谱技术检测数据是指连续采集的液相色谱-质谱联用技术检测的一级谱图数据。
本发明的分析系统中构建的深度学习模型,可以根据用户的需求进行搭建。
本发明基于深度学习模型的尿液蛋白质组学谱图数据分析系统通过“特征刈痕提取”方法对尿液蛋白质组的原始谱图数据进行一致性处理,有效提高了不同样本的谱图数据之间的可比性,克服了本领域的技术难题。
实验证明,利用本发明基于深度学习模型的尿液蛋白质组学谱图数据分析系统能够对尿液蛋白质组的原始lc-ms/ms谱图数据进行解析和识别,从而高准确度的预测该尿液样本对应的对象是健康人还是某种疾病的患者。本发明基于深度学习模型的尿液蛋白质组学谱图数据分析系统在开发尿蛋白异常相关疾病的辅助诊断工具中具有广阔的应用前景。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
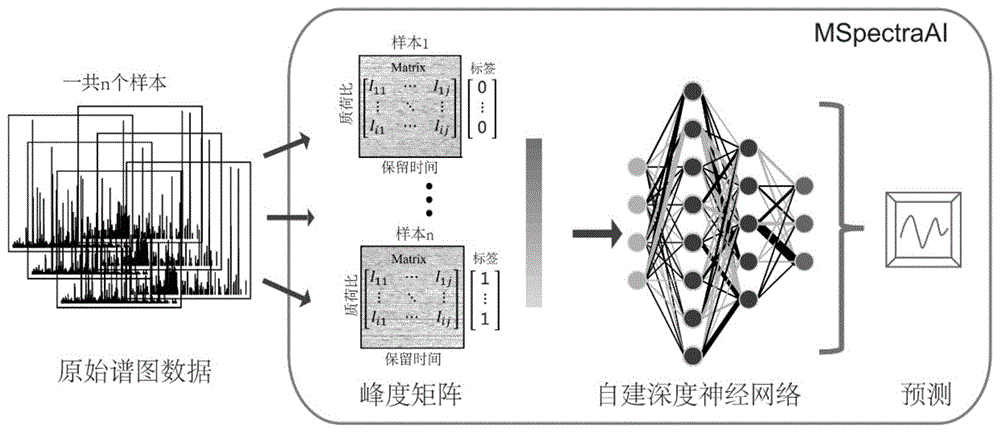
图1是本发明基于深度学习模型的尿液蛋白质组学谱图数据分析系统的运行流程框架示意图。
图2是利用“特征刈痕提取”的方法对原始lc-ms/ms谱图数据进行一致性处理的流程图。
图3是本发明中深度神经网络模型的训练和预测示意图。
具体实施方式
本发明所用原料与设备均为已知产品,通过购买市售产品所得。
实施例1基于深度学习模型的尿液蛋白质组学谱图数据分析系统
本发明所要研究的对象是基于液相色谱-串联质谱技术(简称lc-ms/ms)获得的蛋白质组学原始谱图数据,生物样本为人的尿液。
本发明的分析系统分为三个组成部分(如图1所示),具体如下:
第一部分:一致性处理部分,在该部分对原始lc-ms/ms谱图数据进行一致性处理,得到标准化的峰度矩阵
通过液相色谱-串联质谱仪得到原始谱图数据,因为每一个谱图数据内包含的峰的个数和强度都不一样,所以此类数据不适用于本发明的深度学习模型进行解析,必须要对原始谱图数据进行一致性处理。本发明开发了一种叫做“特征刈痕提取”(featureswathextraction)的方法对原始lc-ms/ms谱图数据进行一致性处理,算法图解如图2所示,具体处理步骤如下:
(a)提取出n个一级原始谱图数据,顺序编号,一级原始谱图的序号依次记为1,2,3,……,n-1,n。然后根据样本的类别对每一个一级原始谱图进行标签标记,比如,若该一级原始谱图数据来源于健康人的样本,则标签标记为“0”,若该一级原始谱图数据来源于轻度患者的样本,则标签标记为“1”,若该一级原始谱图数据来源于重度患者的样本,则标签标记为“2”。
(b)预先设置“刈痕”的数量,这里的一个刈痕指的是一级原始谱图数据中一小段质荷比的范围,也可以称为一个窗口。然后根据设置的刈痕数量自动算出每个窗口的范围,比如在采集的一级原始谱图数据中,一级的质荷比范围为[300,1400],预先设置刈痕的数量为400,即每一个谱图数据都分为400个窗口,那么本发明的系统会自动根据这个设置计算得到每个窗口的范围(即[300,302.75],[302.75,305.5],……,[1397.25,1400])。每一个窗口的范围计算好以后,本发明的系统会将每一个窗口的谱图数据按照以下公式(1)进行叠加,这样就能保证每一个窗口中只有一个数值:
其中im是指intensitymatrix,即峰度矩阵,i表示谱图数据的序号,j表示窗口的序号,k表示第i个谱图数据、第j个窗口中峰的序号,
(c)峰值标准化。经过步骤(b)的叠加后,每一个谱图数据的异质性仍然较大,即每个谱图数据之间还没有一个统一的尺度,因此可比性较低。本发明的系统利用公式(2)先将每一个谱图数据中每一个窗口的峰值之和分别除以该谱图数据中的最大峰值,然后得到标准化后的峰度矩阵(nim),这样就能使数据之间更具可比性。
nim=im/(max(i1,1…j),…,max(ii,1…j))(2)
其中nim表示normalizedintensitymatrix,即标准化后的峰度矩阵,max(ii,1…j)表示序号为i的谱图数据中最大的峰值,im表示步骤(b)计算得到的峰度矩阵。
第二部分:深度学习模型构建部分
本发明使用的深度学习模型是深度神经网络模型,其基于keras框架搭建,其内嵌了一个初级的深度神经网络模型(由以下部分组成:输入层、3个隐藏层(其中第一层128个节点、第二层64个节点、第三层32个节点)、输出层),隐藏层中激活函数为线性整流函数relu,输出层激活函数为归一化指数函数softmax。
第三部分:模型的训练和预测部分
将第一部分进行一致性处理后的数据分为训练集和测试集(训练和测试样本均为独立样本,相互不交叉),先用训练集数据对第二部分构建的深度神经网络模型进行训练,最后再用训练后的深度神经网络模型对测试集数据对应尿液样本的类别进行预测,并检查深度神经网络模型预测结果的准确度。
为了验证本发明上述分析系统的预测准确性,使用“留一法”的策略对深度神经网络模型的预测能力进行评估(过程如图3所示)。具体操作如下:收集人的尿液样本的n个蛋白质组学原始lc-ms/ms数据,其中来源于健康人的数据标签标记为“0”,来源于轻度患者的数据标签标记为“1”,来源于重度患者的数据标签标记为“2”。然后在每一次循环中,将n-1个经过第一部分的一致性处理后的数据作为训练集,对第二部分构建的深度神经网络模型进行训练,剩下的1个经过第一部分的一致性处理后的数据作为测试集数据。模型训练好以后对该测试集数据进行预测,如此循环n次,就可以得到每一个数据的预测结果,将该预测结果与最初的数据标签标记进行对比,就可以统计出预测结果的总正确率。
以下进一步说明如何预测每一个数据对应的样本的类别。如图3b所示,在每一次迭代过程中,比如要预测第k个数据对应的样本的类别,则需要先将其他n-1个蛋白质组学原始lc-ms/ms数据标准化后的峰度矩阵和对应的类别标签(健康人的为“0”,轻度患者的为“1”,重度患者的为“2”)输入给第二部分预先构建好的深度神经网络模型,该模型可以根据每一类别的数据特征进行训练,得到训练好的模型。接下来,将第k个数据标准化后的峰度矩阵输入到上述训练好的模型,该模型就可以对第k个数据中的每一个谱图数据的类别进行预测,那么,每一个谱图数据都可以得到3个概率值:类别为“0”的概率值,类别为“1”的概率值,类别为“2”的概率值。然后系统会自动将3个概率值中的最大值对应的类别标签作为该谱图数据类别的预测结果。预测完以后,将预测的类别与原始的类别标签进行比较,预测正确的画红勾,预测错误的画红叉,然后统计类别预测正确的谱图数据个数占总谱图数据个数的比例,如果该比例在3个类别结果中最大,则表示对第k个数据对应的样本类别预测正确,否则表示预测错误。
采用上述相同的方法,对其余n-1个数据对应的样本的类别进行循环预测,最后统计预测正确的样本个数占所有样本个数的比例,并将该比例作为评价参数,评价本发明分析系统的预测准确度。比如,若该比例大于等于0.8,则认为本发明的方法预测效果良好,如果该比例小于0.8,则认为本发明的方法预测效果不佳。
以下通过实验例证明本发明的有益效果。
实验例1本发明基于深度学习模型的尿液蛋白质组学谱图数据分析系统的预测效果评估
采用本发明基于深度学习模型的尿液蛋白质组学谱图数据分析系统,对4个健康人的尿液样本、6个受新型冠状病毒(sars-cov-2)感染的病人(其中3个轻度感染、3个重度感染)尿液样本、2个康复病人的尿液样本(康复病人来源于上述6个受新型冠状病毒感染的病人中的2个)的一级原始lc-ms/ms谱图数据的类别进行预测(测试数据来源于公共数据库iprox,数据id:ipx0002166000),结果如表1所示。
表1本发明基于深度学习模型的尿液蛋白质组学原始谱图数据分析系统的效果评估
表1中,原始标签标记和预测类别中,“0”表是健康人,“1”表示受新型冠状病毒感染的轻度患者,“2”表示受新型冠状病毒感染的重度患者,11号样本由5号病人康复后得到,12号样本由10号病人康复后得到。其中轻度和重度的诊断标准是依据中国人民解放军总医院第五医疗中心印发的《新型冠状病毒肺炎的诊断与管理方案(试行第六版)》,比如轻度感染患者主要表现为发热、非肺炎或轻度肺炎,而重度患者主要表现为呼吸困难,呼吸频率≥30次/分钟,平均氧饱和度(≤93%,静息状态)或动脉血氧分压/氧浓度(pao2/fio2≤300mmhg),和/或24-48小时内肺浸润>50%。
预测正确率1是指预测健康人和不同程度的患者(轻度患者和重度患者视为两类)之间的准确度,预测正确率2是指预测健康人和患者(轻度健康和重度健康被统一视为一类)之间的准确度。
从表1可以看出,采用本发明基于深度学习模型的尿液蛋白质组学谱图数据分析系统,可以对人尿液蛋白质原始lc-ms/ms谱图数据进行高准确度的预测。以1-1~10-2样本的数据作为原始lc-ms/ms谱图数据库,健康人和患者之间的预测准确度为100%,健康人及不同程度的患者之间预测准确度为85%。进一步地,以1-1~10-2样本的数据作为训练数据,以11-1~12-2样本的数据作为测试数据,发现患者康复以后,本发明的分析系统也可以对其进行准确的预测(预测准确度为100%),从而准确的识别该尿液样本对应的对象是健康人还是尚处于感染期的新型冠状病毒感染患者。
综上,本发明提供了一种基于深度学习模型的尿液蛋白质组学谱图数据分析系统。该分析系统利用“特征刈痕提取”方法对尿液蛋白质组的原始谱图数据进行一致性处理,有效提高了不同样本的谱图数据之间的可比性,克服了本领域的技术难题。实验证明,本发明的分析系统能够对尿液蛋白质组的lc-ms/ms谱图数据进行解析和识别,从而高准确度的预测该尿液样本的来源是健康人还是疾病患者。该分析系统在开发与尿蛋白异常相关疾病的辅助诊断工具中具有广阔的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!