血浆中丙泊酚的快速固相萃取检测方法与流程
本发明涉及一种血浆中丙泊酚浓度的快速固相萃取检测方法。
背景技术:
丙泊酚被广泛用于诱导和维持麻醉。丙泊酚因其在快速诱导和消除、效果持续时间短、麻醉后恢复温和、副作用少以及未发现致畸作用方面的卓越表现,而通常被认为是最适合短期镇静的静脉麻醉药。在先前的研究报道中,细胞色素p450酶的基因型、患者的体重指数(bmi)及年龄等与丙泊酚的药代动力学参数具有相关性的影响因素导致丙泊酚麻醉存在显著的个体间差异且频繁被报道。除医疗用途外,滥用丙泊酚引起的成瘾和死亡在世界范围内也非常广泛。使用过高剂量或过低剂量丙泊酚可分别引起丙泊酚输注综合征和术中知晓的发生,从而给患者带来生理和心理伤害。综上所述,血浆丙泊酚浓度的检测对于药物代谢动力学以及法医毒理学研究是非常重要的。
气相色谱-质谱联用(gc-ms)和液相色谱-质谱联用(lc-ms)是一种特异性、灵敏度和准确度高的新技术,常被应用于丙泊酚血浆浓度的监测。血浆需经过样品前处理之后,才能用于液相色谱-质谱联用分析。固相萃取(solid-phaseextraction,spe)利用c18硅胶色谱填料选择性吸附提取血浆基质中非极性的目标化合物,是在质谱分析之前用于样品的分离,浓缩和纯化目的的常规样品前处理方法。但是,传统的固相萃取方法需先对血浆样品进行蛋白沉淀或液液提取等预处理过程,整个操作耗时费力且相关耗材成本较高,需使用固相萃取仪等专用仪器设备,且对操作人员有一定的技术要求,大大限制了其在丙泊酚分析检测中的广泛应用研究。
传统的固相萃取方法如果不对血浆样品进行蛋白沉淀或液液提取等预处理过程,往往会造成固相萃取柱的堵塞,导致固相萃取后续操作无法完成。
技术实现要素:
本发明要解决的技术问题是提供一种使用方便的血浆中丙泊酚浓度的快速固相萃取检测方法。
为了解决上述技术问题,本发明提供一种血浆中丙泊酚浓度的快速固相萃取检测方法,包括以下步骤:
1)、c18移液器吸头固相萃取:
c18移液器吸头内含有c18硅胶填料,包括以下步骤:
1.1)、先利用50%乙腈水溶液湿润并激活(充分湿润并激活)吸头内的c18硅胶填料,再利用1%甲酸水溶液平衡吸头内的c18硅胶填料,得预处理后的c18移液器吸头;
1.2)、在1ml待测血浆中加入10μl50μg/ml的丙泊酚-d17内标溶液,调节ph至(4±0.2),得血浆样品;
1.3)、取100μl血浆样品,利用步骤1.1)所得的预处理后的c18移液器吸头对其进行反复吸入并排出的操作;
1.4)、先用0.1%甲酸水溶液对步骤1.3)结束后的吸头内的c18硅胶填料进行冲洗;
再以甲醇为洗脱溶剂,对吸头内的c18硅胶填料进行洗脱,得样品溶液;
1.5)、将样品溶液于(8±1)℃离心(10000×g离心5分钟),离心所得的上清液(约50μl)进行后续的液相色谱-质谱分析;
说明:离心所得的上清液移入样品瓶并在液相色谱-质谱分析之前于-80℃下储存。
2)、液相色谱-质谱分析;
3)、将步骤2)所得的丙泊酚峰面积与丙泊酚-d17峰面积的比值作为x,带入y=0.0588x+0.0053,y为待测血浆中丙泊酚的浓度,单位为μgml-1。
作为本发明的血浆中丙泊酚的快速固相萃取检测方法的改进,所述步骤2)中:
2.1)液相色谱分离:
上清液的进样量为5μl,在分析过程中,柱温保持在30℃;
洗脱溶剂由洗脱溶剂a和洗脱溶剂b组成,洗脱溶剂a为0.1%甲酸水溶液,洗脱溶剂b为乙腈,线性梯度洗脱的流速设置为0.4mlmin-1;
色谱分离条件如下:
0.0分钟~0.5分钟,20%a~5%a;
0.5分钟~2.0分钟,5%a;
2.0分钟~2.1分钟,5%a~20%a;
2.1分钟~4.0分钟,20%a;
2.2)、质谱分析:
大气压化学电离(apci)被用作分析丙泊酚和丙泊酚-d17的离子源;
设定参数如下:离子源温度为150℃,毛细管电压-4.5kv,去溶剂化温度500℃,去溶剂化气体流量800l/h,锥孔气体流量50l/h;三重四极杆质谱仪以选择反应监测srm模式运行;m/z177→m/z177用于丙泊酚监测;m/z194→m/z194用于丙泊酚-d17监测;两个通道的相应锥电压和碰撞能均设置为46v和18v;
分别得到上清液的丙泊酚峰面积和丙泊酚-d17峰面积。
作为本发明的血浆中丙泊酚的快速固相萃取检测方法的进一步改进,步骤2)中:
使用c18反相超高效液相色谱柱(hsst31.8μm,100mm×2.1mm)的watersacquityh-class超高效液相色谱系统进行液相色谱分离,将负离子化模式下的沃特世xevotqd三重四极杆质谱仪(英国沃特世公司)与液相色谱系统联合进行质谱分析。
作为本发明的血浆中丙泊酚的快速固相萃取检测方法的进一步改进:
步骤1.3)中反复吸入并排出的操作次数为5~7次。
作为本发明的血浆中丙泊酚的快速固相萃取检测方法的进一步改进:
步骤2.1)中,进样温度为8℃。
作为本发明的血浆中丙泊酚的快速固相萃取检测方法的进一步改进:
c18移液器吸头为安捷伦omixc18移液器吸头(10-100μl);
步骤1.1)中:先吸入100μl50%乙腈的水溶液再排出废液,然后重复上述吸入50%乙腈的水溶液再排出废液的动作1~3次(即,共操作2~4次);从而实现充分湿润并激活吸头内的c18硅胶填料;
接着,先吸入100μl1%甲酸水溶液再排出废液,然后重复上述吸入1%甲酸水溶液、排出废液的动作1~3次(即,共操作2~4次),从而实现平衡移液吸头内的c18硅胶填料。
作为本发明的血浆中丙泊酚的快速固相萃取检测方法的进一步改进:
步骤1.2)中:利用甲酸调节ph值。
作为本发明的血浆中丙泊酚的快速固相萃取检测方法的进一步改进:
步骤1.4)中:先吸入100μl0.1%甲酸水溶液,再排出废液,然后重复上述吸入0.1%甲酸水溶液再排出废液的动作1~3次(即,共操作2~4次);从而实现用0.1%甲酸水溶液对步骤1.3)结束后的吸头内的c18硅胶填料进行冲洗;
使用50μl甲醇作为洗脱溶剂,吸头吸入后再排出,对排出液重复进行吸头吸入后再排出的动作1~3次,最后一次的排出液最终提取的样品溶液。
在本发明中,使用手持移液器,利用玻璃纤维支撑填充固定体积或重量的c18硅胶等非极性色谱分离材料的移液器吸头,通过吸取和排出不同液体,完成整个c18移液器吸头固相萃取过程。第一步,依次使用50%乙腈水溶液和1%甲酸水溶液对c18硅胶等色谱料填料进行活化和平衡;还包括制备血浆样品;第二步,利用c18硅胶等色谱填料选择性吸附提取血浆中的非极性的丙泊酚分子,可反复操作提高丙泊酚的提取效率;第三步,通过吸取和排出0.1%甲酸水溶液过程,洗去在c18硅胶等色谱填料内的吸附较差或不吸附的的极性基质分子;第四步,利用甲醇有机溶剂洗脱c18硅胶等色谱填料吸附的丙泊酚分子,用于随后的气相色谱-质谱分析或液相色谱-质谱分析。
本发明利用填充少量c18硅胶色谱填料的移液吸头,无需对血浆样品进行蛋白沉淀等额外的提取过程,直接从血浆样品进行固相萃取,分离纯化非极性的丙泊酚药物分子,用于气相色谱-质谱联用分析或液相色谱-质谱联用。
本发明与传统的固相萃取法相比,c18移液吸头固相萃取法省去了血浆的蛋白沉淀等血浆样品的预处理过程,耗材成本低,仅使用实验室常见的手持移液器,不需要额外的仪器设备(固相萃取仪),即可完成整个固相萃取过程。本发明的色谱分离时间仅仅需要4分钟,低于常规的色谱分离方法所需时间(5分钟以上),本发明使用了与丙泊酚化学性质更接近的丙泊酚-d17作为内标,能提高检测结果的准确度和精密度。
综上所述,本发明的c18移液吸头固相萃取法耗材成本低、快速、操作简单、易行,技术难度低,提高了分析检测效率,适用范围广,可在临床药代动力学和法医毒理学等领域得到广泛应用。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细说明。
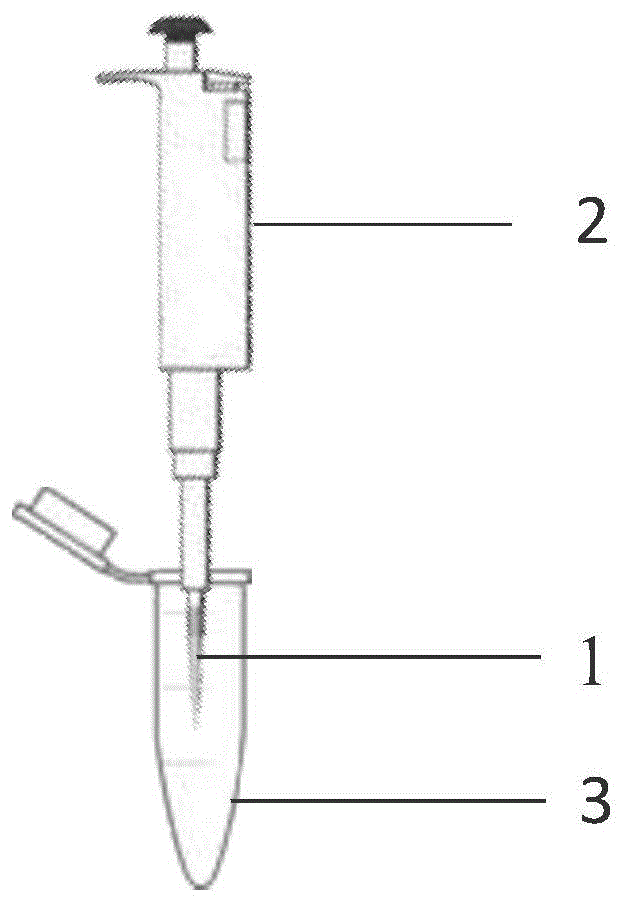
图1是c18移液吸头固相萃取示意图;
图1中,1:c18移液器吸头;2:手持移液器;3:血浆;
图2是丙泊酚(a)和丙泊酚-d17(b)的化学结构和二级碎裂质谱图谱。
图3是各血浆的srm监测图谱;
图3中,
(a)、空白人血浆;(b)、0.005μgml-1丙泊酚的人血浆标准品;(c)0.5μgml-1丙泊酚的人血浆标准品;(d)丙泊酚麻醉受试者血浆;
除了(a)之外,(b)~(d)这3种血浆样品中均含有0.5μgml-1丙泊酚-d17内标。
图4为人血浆丙泊酚浓度检测标准曲线;
图5为丙泊酚靶控输注麻醉病人血浆丙泊酚浓度的实验测定。
具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
实施例1:
1.c18移液吸头固相萃取方法
手持移液器插入安捷伦omixc18移液器吸头(10-100μl,美国密理博公司),先吸入100μl50%乙腈的水溶液再排出废液,然后重复上述吸入50%乙腈的水溶液再排出废液的动作2次(即,共操作三次),以充分湿润并激活吸头内的c18硅胶填料。随后,先吸入100μl1%甲酸水溶液再排出废液,然后重复上述吸入1%甲酸水溶液、排出废液的动作2次(即,共操作三次),以平衡移液吸头内的c18硅胶填料。
在1ml待测血浆中加入10μl50μg/ml的丙泊酚-d17内标溶液,并加入约4μl甲酸将ph值调节到4,得血浆样品;取100μl血浆样品用上述激活并平衡后的c18移液器吸头、以同一个离心管作为接收容器,反复吸入并排出(操作次数共6次),以提高丙泊酚的提取效率。
为了冲洗移液器吸头内的吸附血浆丙泊酚的c18硅胶填料,吸入100μl0.1%甲酸水溶液,再排出废液,然后重复上述吸入0.1%甲酸水溶液再排出废液的动作2次(操作次数共3次)。
最后,使用50μl甲醇作为洗脱溶剂,以同一个离心管作为接收容器,反复吸入并排出(操作次数共3次),位于离心管中的最后一次的排出液为最终提取的样品溶液。
将该样品溶液在8℃以10000×g离心5分钟,将所得的上清液(约50μl)移入样品瓶并在液相色谱-质谱分析之前于-80℃下储存。
2.色谱分离方法
通过使用c18反相超高效液相色谱柱(hsst31.8μm,100mm×2.1mm)的watersacquityh-class超高效液相色谱系统进行液相色谱分离,在分析过程中,柱温保持在30℃。
洗脱溶剂由洗脱溶剂a和洗脱溶剂b组成,洗脱溶剂a为0.1%甲酸水溶液,洗脱溶剂b为乙腈,线性梯度洗脱的流速设置为0.4mlmin-1。
最终优化的色谱分离条件如下:
0.0分钟~0.5分钟,20%a~5%a;
0.5分钟~2.0分钟,5%a;
2.0分钟~2.1分钟,5%a~20%a;
2.1分钟~4.0分钟,20%a。
自动进样器温度设置为8℃,在分析之前如上所述临时存储样品,样品溶液的进样量设置为5μl。
3.质谱分析方法
将负离子化模式下的沃特世xevotqd三重四极杆质谱仪(英国沃特世公司)与液相系统联用进行质谱分析。大气压化学电离(apci)被用作分析丙泊酚和丙泊酚-d17的离子源。
具体仪器参数如下所示:离子源温度为150℃,毛细管电压-4.5kv,去溶剂化温度500℃,去溶剂化气体流量800l/h,锥孔气体流量50l/h。三重四极杆质谱仪以选择反应监测srm模式运行。做完丙泊酚和丙泊酚-d17的二级碎裂质谱图谱后(图2),最终确定两个单独通道:m/z177→m/z177用于丙泊酚监测;m/z194→m/z194用于丙泊酚-d17监测。两个通道的相应锥电压和碰撞能均设置为46v和18v。
实施例2、方法的特异性验证
为了研究丙泊酚检测方法的特异性,收集了来自6个72小时内未曾使用过丙泊酚的健康受试者的空白人血浆样品,按照实施例1步骤1~步骤3所述的使用c18移液吸头固相萃取方法直接对血浆样品进行处理(未加入丙泊酚-d17内标物),然后进行液相色谱-质谱分析。
上述空白人血浆样品进行检测时,丙泊酚峰面积为0;丙泊酚-d17峰面积为0。
即,如图3a所示,在用于丙泊酚和丙泊酚-d17监测的两个选择反应监测srm通道中均未检测到血浆基质的明显影响。
实施例3、方法灵敏度验证
将丙泊酚和固定浓度的丙泊酚-d17内标溶液加入空白人血浆样品后,配制成丙泊酚浓度为0.005μgml-1人血浆标准品(丙泊酚-d17内标浓度0.5μgml-1),然后用实施例1的步骤1~步骤3所述方法进行样品处理和液相色谱-质谱联用分析。
丙泊酚监测通道m/z177→m/z177,丙泊酚峰面积为6,信噪比为10。
丙泊酚-d17监测通道m/z194→m/z194,丙泊酚-d17峰面积为703。
所得结果如图3b所示,定量下限lloq确定为0.005μgml-1。
实施例4、方法的线性、准确度和精密度验证
将不同浓度的丙泊酚和固定浓度的丙泊酚-d17内标溶液加入空白人血浆样品后,配制成丙泊酚浓度为0.005、0.01、0.5、0.1、0.5、1.0、2.0和5.0μgml-1的人体血浆校准标准品(丙泊酚-d17内标浓度0.5μgml-1),然后用实施例1的步骤1~步骤3所述方法进行样品处理和液相色谱-质谱联用分析,测试定量方法的线性。
如图4所示,使用加权最小二乘回归方法通过线性回归模型构建校正曲线。使用1/x作为权重因子校准曲线的平均线性回归方程表示为y=0.0588x+0.0053,其中y表示血浆丙泊酚浓度,x表示丙泊酚与丙泊酚-d17内标的锋面积之比。线性相关系数r2为0.9992,证明方法的线性较好。人血浆标准品中丙泊酚(0.5μgml-1)和丙泊酚-d17(0.5μgml-1)的代表性选择反应监测srm图谱如图3c所示。
丙泊酚监测通道m/z177→m/z177的定量结果为:0.4839μgml-1
即,0.5μgml-1浓度时,丙泊酚峰面积为5226;丙泊酚-d17峰面积为642。
如表1所示,使用加入最低定量限浓度丙泊酚(0.005μgml-1)和加入低浓度(0.015μgml-1)、中度(0.3μgml-1)、高浓度(3.0μgml-1)丙泊酚的人血浆样品评估了方法的准确性和精密度。在每个评估中重复五次。最后,批次内准确度确定为90.0-108.6%,批次间准确度确定为93.3-113.3%。所有这些结果均满足生物分析方法的要求的85-115%范围。同时,批次内精密度和批次间精密度分别确定为2.0-8.9%和3.8-7.7%,满足±15%的标准。除了准确性和精密度外,还对三种不同浓度水平的质量控制样品进行了回收率和稳定性的研究,发现其在生物分析方法要求的85-115%范围内。
表1、准确度和精密度结果
实验1、
在临床实践中,较低或较高浓度的血浆丙泊酚可能分别导致术中意识或心肺停止。因此测定血浆丙泊酚浓度在麻醉中非常重要。经当地伦理委员会批准,并获得知情同意。由医院提供实施丙泊酚全身麻醉的受试者血浆,对血浆丙泊酚浓度进行实验测定,具体实验方法如下:
1、c18移液吸头固相萃方法
同实施例1的步骤1。
2、色谱分离方法
同实施例1的步骤2。
3、质谱分析方法
同实施例1的步骤3。
分别获得丙泊酚峰面积和丙泊酚-d17峰面积,计算丙泊酚和丙泊酚-d17的峰面积比值作为x,带入校正曲线y=0.0588x+0.0053,所得y为待测血浆(受试者血浆)的丙泊酚浓度。
所得结果如下表2所述。
表2
对比实验1、将实验1中的洗脱溶剂由甲醇改成“甲醇:水=80:20,v/v”,体积用量保持不变;并代入该洗脱溶剂所对应的校正曲线中,其余等同于实验1。
仅仅对表2待测血浆a、待测血浆f进行检测,所得结果为:
待测血浆a的丙泊酚浓度为1.88μgml-1,待测血浆f的丙泊酚浓度为0.20μgml-1。该方法的准确度和回收率都变差。
对比实验2、将实验1中用于“充分湿润并激活吸头内的c18硅胶填料”的“50%乙腈的水溶液”改成“甲醇”,用量保持不变;并代入该实验条件所对应的校正曲线中,其余等同于实验1。
仅仅对表2待测血浆a、待测血浆f进行检测,所得结果为:
待测血浆a的丙泊酚浓度为2.01μgml-1,待测血浆f的丙泊酚浓度为0.26μgml-1。该方法的准确度和回收率都变差。
对比实验3、将本实验1采用的血浆样品直接用常规固相萃取方法处理,会导致固相萃取小柱堵塞,进而无法进行后续实验操作,无法得出丙泊酚浓度的分析检测结果。
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!