一种测定盐酸氯丙嗪中有关物质的方法与流程
本发明涉及一种药品的检测方法,特别涉及一种测定盐酸氯丙嗪中有关物质的方法。
背景技术:
1、盐酸氯丙嗪,别名:2-氯-10-(3-二甲氨基丙基)吩噻嗪盐酸盐,cpz,氯普马嗪,分子式:c17h19cln2s.hcl,cas号:69-09-0,分子量:355.33,为一种吩噻嗪类抗精神病药,结构式如下:
2、
3、其作用机制主要与其阻断中脑边缘系统及中脑皮层通路的多巴胺受(da2)有关。对多巴胺(da1)受体、5-羟色胺受体、m-型乙酰胆碱受体、α-肾上腺素受体均有阻断作用,作用广泛。盐酸氯丙嗪片是常用的一种剂型,适应症为1.对兴奋躁动、幻觉妄想、思维障碍及行为紊乱等阳性症状有较好的疗效。2.止呕,各种原因所致的呕吐或顽固性呃逆。
4、中国药典2020年版收载的盐酸氯丙嗪有关物质项检测方法为液相色谱等度方法,无已知杂质的控制;usp中收载的盐酸氯丙嗪片与盐酸氯丙嗪原料均未对有关物质进行控制;ep收载的盐酸氯丙嗪中共有已知杂质6个,分别为杂质a、b、c、d、e、f,
5、结构式分别为:
6、杂质a:
7、
8、c17h19cln2os 334.86
9、杂质b:
10、
11、c21h30cl3n3s 462.91
12、杂质c:
13、
14、c17h21cln2s 320.88
15、杂质d:
16、
17、c16h18cl2n2s 341.30
18、杂质e:
19、
20、c12h8clns 233.72
21、杂质f:
22、
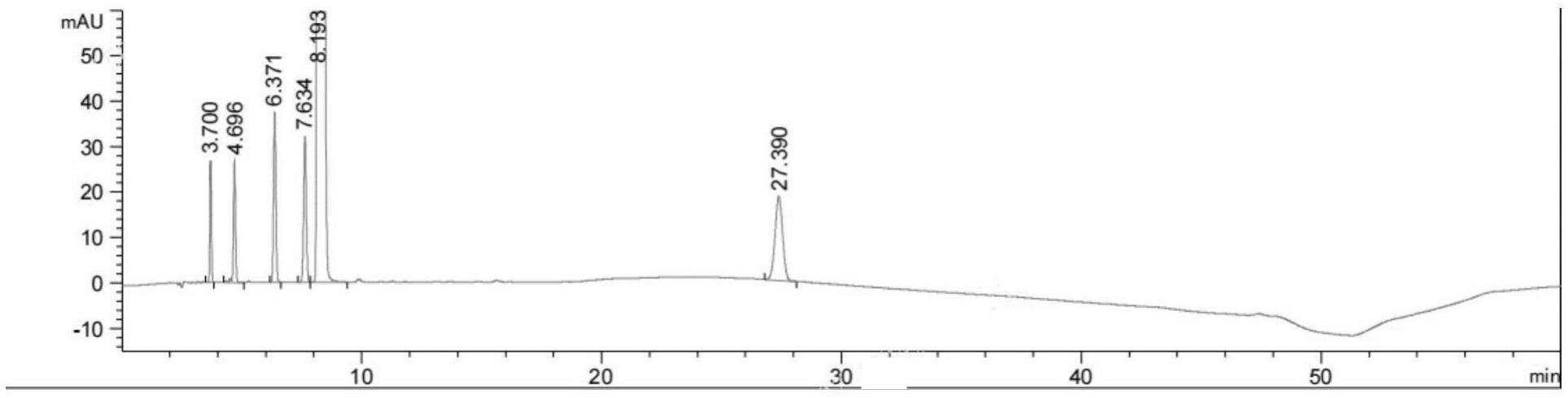
23、c17h19cln2s 318.86
24、其中杂质f为主成分异构体,以单独液相正相方法检测,其余5个杂质均在有关物质项下以液相反相等度方法检测,色谱条件与中国药典2020年半收载的方法一致。
25、参考中国药典的色谱条件,盐酸氯丙嗪保留时间约为10分钟,记录色谱至保留时间的4倍,采集时间约为40分钟,经研究发现将运行方法增加至90分钟时,在约70分钟处发现一个未知杂质峰,且由于保留时间过长,该峰被拉宽,峰高较低,无法准确积分,。因此需重新开发一个梯度洗脱方法,以满足杂质检测强需求。
技术实现思路
1、本发明的目的是提供一种检测盐酸氯丙嗪中有关物质的方法。
2、本发明所述方法,步骤如下:
3、步骤1,对照品溶液的配制:称取盐酸氯丙嗪杂质a、杂质b、杂质c、杂质d及杂质e,用溶剂溶解并稀释至刻度,作为储备液;称取对照品盐酸氯丙嗪,加储备液,用溶剂溶解并稀释至刻度;
4、步骤2,供试品溶液的配制:;称取盐酸氯丙嗪,加溶剂溶解并稀释至刻度;
5、步骤3,测定:取对照品溶液和供试品溶液,注入hplc色谱仪,得到色谱图,根据色谱图,计算供试品溶液中各组分的含量。
6、其中测定过程中的色谱条件如下:
7、色谱柱:以辛烷基硅烷键合硅胶为填充剂[alltima c8(4.6mm×250mm,5μm)];流动相:以0.5%三氟乙酸(用四甲基乙二胺调节ph值至5.3)-乙腈(90:10)为流动相a,以0.5%三氟乙酸(用四甲基乙二胺调节ph值至5.3)-乙腈(10:90)为流动相b,流速为1.0ml/min,柱温为30℃,检测波长为254nm。按下表进行线性梯度洗脱,
8、梯度洗脱条件
9、
10、其中,流动相的配制方法如下:
11、流动相a的配制方法:精密移取三氟乙酸5ml置1000ml水中混匀,用四甲基乙二胺调节ph值至5.3抽滤,量取上述溶液900ml和经微孔滤膜抽滤的乙腈100ml,混匀,超声脱气;流动相b的配制方法:精密移取三氟乙酸5ml置1000ml水中混匀,用四甲基乙二胺调节ph值至5.3抽滤,量取上述溶液100ml和经微孔滤膜抽滤的乙腈900ml,混匀,超声脱气。
12、其中,步骤1,对照品溶液的配制方法如下:精密称取盐酸氯丙嗪杂质a、杂质b、杂质c、杂质d及杂质e各4mg置100ml量瓶中,用溶剂溶解并稀释至刻度,摇匀,作为储备液;精密称取对照品盐酸氯丙嗪对照品40mg,置100ml量瓶中,精密移入10ml储备液,用溶剂稀释至刻度,摇匀,即制成每1ml含盐酸氯丙嗪0.4mg、各杂质4μg的溶液。精密量取对照溶液2ml,置20ml量瓶中,用溶剂稀释至刻度,摇匀。
13、其中,步骤2,供试品溶液的配制方法如下:精密称取盐酸氯丙嗪40mg,置100ml量瓶中,加溶剂溶解并稀释至刻度,摇匀,使用针头式过滤器滤过,得到续滤液1;精密移取1ml经滤过的供试品溶液,置200ml量瓶中,用溶剂稀释至刻度,摇匀,使用针头式过滤器滤过,得到续滤液2。
14、其中,步骤1和步骤2中,所述溶剂为三氟乙酸,四甲基乙二胺,乙腈的混合水溶液,其配制方法如下:精密移取三氟乙酸5ml置1000ml水中混匀,用四甲基乙二胺调节ph值至5.3抽滤,量取上述溶液500ml和经微孔滤膜抽滤的乙腈500ml,混匀,超声脱气。
15、其中,测定过程中各组分的计算采用到相对保留时间与校正因子,其关系如下
16、杂质相对保留时间
17、
18、本发明最优选的方法,步骤如下:
19、步骤1,对照品溶液的配制:精密称取盐酸氯丙嗪杂质a、杂质b、杂质c、杂质d及杂质e各约4mg置100ml量瓶中,用溶剂溶解并稀释至刻度,摇匀,作为储备液;
20、精密称取对照品盐酸氯丙嗪对照品约40mg,置100ml量瓶中,精密移入10ml储备液,用溶剂稀释至刻度,摇匀,即制成每1ml含盐酸氯丙嗪0.4mg、各杂质4μg的溶液。精密量取对照溶液2ml,置20ml量瓶中,用溶剂稀释至刻度,摇匀。
21、步骤2,供试品溶液的配制:精密称取盐酸氯丙嗪约40mg,置100ml量瓶中,加溶剂溶解并稀释至刻度,摇匀,使用针头式过滤器滤过,得到续滤液1;精密移取1ml经滤过的供试品溶液,置200ml量瓶中,用溶剂稀释至刻度,摇匀,使用针头式过滤器滤过,得到续滤液2。
22、步骤3,测定:取对照品溶液和供试品溶液,注入hplc色谱仪,得到色谱图,根据色谱图,计算供试品溶液中各组分的含量。
23、其中,步骤1和步骤2中,所述溶剂为三氟乙酸,四甲基乙二胺,乙腈的混合水溶液,其配制方法如下:精密移取三氟乙酸5ml置1000ml水中混匀,用四甲基乙二胺调节ph值至5.3抽滤,量取上述溶液500ml和经微孔滤膜抽滤的乙腈500ml,混匀,超声脱气(当配制量大于1000ml时,其取用量均应按比例增加)。
24、测定过程中的色谱条件如下:
25、色谱柱:以辛烷基硅烷键合硅胶为填充剂[alltima c8(4.6mm×250mm,5μm)],流动相:以0.5%三氟乙酸(用四甲基乙二胺调节ph值至5.3)-乙腈(90:10)为流动相a,以0.5%三氟乙酸(用四甲基乙二胺调节ph值至5.3)-乙腈(10:90)为流动相b,
26、流速为1.0ml/min,柱温为30℃,检测波长为254nm。
27、按下表1进行线性梯度洗脱,
28、表1、梯度洗脱条件
29、
30、测定过程中各组分的计算采用到相对保留时间与校正因子,其关系如下
31、表2、杂质相对保留时间
32、
33、其中,流动相的配制方法:
34、流动相a的配制方法:精密移取三氟乙酸5ml置1000ml水中混匀,用四甲基乙二胺调节ph值至5.3抽滤,量取上述溶液900ml和经微孔滤膜抽滤的乙腈100ml,混匀,超声脱气(当配制量大于1000ml时,其取用量均应按比例增加)。
35、流动相b的配制方法:精密移取三氟乙酸5ml置1000ml水中混匀,用四甲基乙二胺调节ph值至5.3抽滤,量取上述溶液100ml和经微孔滤膜抽滤的乙腈900ml,混匀,超声脱气(当配制量大于1000ml时,其取用量均应按比例增加)。
36、本发明的方法,和现有技术相比,优点如下:
37、梯度洗脱的方法能够缩短主峰后出峰的杂质的保留时间,缩短检测时间,提高检测效率。优化杂质的峰型,使峰高更高,峰型更好看,提高杂质峰的积分准确度,也提高了检测的灵敏度。
- 还没有人留言评论。精彩留言会获得点赞!