小麦BSR‑Seq基因定位的方法与流程
本发明涉及一种遗传育种技术,尤其涉及一种小麦BSR-Seq(Bulked Segregant RNA-Seq,混池转录组测序)基因定位的方法。
背景技术:
小麦是人类主要口粮作物之一,在耕地减少、需求量不断上升的背景下,其产量丰欠攸关粮食安全。培育高产优质抗逆小麦品种和遗传改良小麦重要农艺性状是小麦稳产增产的重要方法。提高小麦传统育种方法的效率、不断地创新和应用新的育种技术是必经之路。当前分子标记技术、标记辅助选择育种和基因组选择愈发得到关注,利用紧密连锁的分子标记鉴定和筛选与表型相关的染色体区段或基因对于基因精细定位和克隆、标记辅助选择育种、基因聚合育种、分子设计育种具有重要意义和应用价值。然而,与小麦重要农艺性状紧密连锁的分子标记的开发面临诸多挑战,主要由于小麦缺乏基因组序列信息且基因组复杂,让基于PCR的分子标记开发较为困难,来源于基因的PCR标记和RFLP标记常扩增或杂交到小麦的部分同源染色体上,因此常被定位到部分同源染色体上。当前有多种方法用于解决部分问题。
第一种方法是利用小麦染色体片段缺失系和缺体-四体系。四百多套中国春染色体片段缺失系得到开发(Endo等,1996),每个系都有一个染色体片段被删除,有大量EST序列锚定在了特定删除区段内(Qi等,2004),这可以通过细胞遗传学的方法进行检测。另外,一系列染色体缺体-四体系也得到开发,每个系的一对染色体被其部分同源的染色体替换。理想的染色体缺体-四体系只在含有目的基因的染色体被替换,由此构建的作图群体使多态性标记定位到期望的染色体上,避免了定位到部分同源染色体上。如Fairs等用染色体片段缺失系和缺体-四体系成功克隆了驯化基因Q,定位中用只在5A染色体Q位点有差异的亲本构建了462个F2个体就将该基因成功克隆(Faris等,2003)。但这种方法所开发的分子标记的数量有限,定位精度较差,因此应用有限。
第二种方法是基于比较基因组学的方法。虽然禾本科物种在基因组大小上差异很大,但基因在染色体上的顺序保持着广泛的保守性,即近缘物种间保守的基因共线性关系,这反映了禾本科物种在五千万年前从同一个祖先种分化而来的事实(Paterson等,2004)。这同时为利用共线性开发分子标记提供了基础。模式禾本科物种水稻和短柄草拥有小的基因组且和小麦保持着良好的共线性关系,他们的参考基因组序列已经完成,为小麦基因的图位克隆提供了理想的参照,故被广泛运用于小麦分子标记的开发当中(Kellogg 2015),如抗条锈病基因Lr67的图位克隆中利用与水稻和短柄草的保守共线性关系快速的找到了目的基因所在的BAC(Moore等,2015),其他很多基因如Sr33、Sr35等都利用了比较基因组学开发分子标记(Periyannan等,2013;Saintenac等,2013)。当定位区间对应的水稻或短柄草基因组区段得到确定后,即可利用共线性区域内的短柄草或水稻基因开发小麦的探针甚至筛选BAC,这提高了基因图位克隆的效率,当前的大量数据分析表明65%左右的短柄草或水稻基因和小麦保持着良好的共线性关系(Brutnell等,2015;Kellogg 2015)。但比较基因组学也有一定的局限性,在一些小麦基因组区段发生了重组破坏了和水稻或短柄草的共线性关系,小麦基因组的扩增增加了很多非共线性的基因(Glover等,2015;International Wheat Genome Sequencing Consortium 2014;Wicker等,2010;Wicker等,2011)。如快速进化的抗病基因常分布在重组率较高的基因组区域,此类区域因重组频繁发生共线性容易遭到了破坏,一些基因组区段甚至找不到对应的水稻或短柄草共线性区间(Leister 2004)。因此禾本科物种间保守的共线性关系为小麦基因的定位提供了参考,但在小麦基因的精细定位、候选基因鉴定中作用有限,特别是在基因组复杂的区段内。
第三种方法是基于芯片技术的方法。基于芯片杂交的分型方法通量高、成本低,在当前挖掘多态性和关联分析中应用广泛。当前有小麦9k(Cavanagh等,2013)、90k(Wang等,2014)和820k(Wilkinson等,2012)芯片,标记密度逐渐增高。然而芯片中的标记是依据特定品种开发而来,对于其他品种的分型效率不高,小麦基因的多拷贝特征使有大量的SNP无信号或并不能分型,同时SNP密度在染色体上分布不均一,大量SNP聚集在少量位点上,另外,其SNP数量固定且定位精度不高,如要对每个个体进行分型则成本较高。因此当前芯片技术虽然可以挖掘大量的变异,但其成本较高且定位精度有限,特别是在小麦重要基因的精细定位和克隆中应用有限。
第四种方法是基于下一代测序技术的方法。下一代测序技术主要以高通量低成本著称,这些特点大大拓展了可研究的范围,比如除了得到DNA序列信息之外,我们可以用这些基于测序的方法来研究基因的表达,高效且准确的获得基因的表达量、稀有转录本、选择性剪切事件、非编码转录本、非编码区、结构变异和单核苷酸多态性等各种信息,这是基因芯片等技术无法比拟的,实际上基于测序的方法正在取代基因芯片成为研究基因表达最有力的工具(Farnham 2009;Licatalosi等,2010;Wang等,2009)。然而,小麦缺乏参考基因组序列,这大大限制了下一代测序技术在小麦重要基因挖掘和定位中的应用。当前可用的方法是利用GBS(Genotyping By Sequencing)技术,其不依赖于参考基因组序列,利用序列的相似性进行聚类和分型,在小麦中有少量应用报道(Edae等,2015;Mascher等,2013)。但该技术获得高质量SNP标记有限,且小麦基因的高拷贝特征容易带来误差,其需要对每个个体进行测序成本较高,因而应用有限。
技术实现要素:
本发明的目的是提供一种不依赖于参考基因组序列、低成本、快速、精度高的小麦BSR-Seq基因定位的方法。
本发明的目的是通过以下技术方案实现的:
本发明的小麦BSR-Seq基因定位的方法,包括步骤:
A、混池的构建和测序:
根据重组自交系作图群体、加倍双单倍体(DH)群体、回交渗入系群体、F2或F2:3分离群体表型鉴定结果,分别用15-30个以上纯合极端高值个体和15-30以上个纯合极端低值个体分别组建高值混合池和低值混合池,在表型未表现出差异,或表现出差异后分别取等量叶片组织混合而成高值池和低值池,并提取高值池和低值池的mRNA后进行转录组测序,从而得到两个混池的转录组测序数据。
B、高质量变异挖掘:
首先,对转录组测序原始数据进行过滤得到高质量数据,过滤标准是去除两端测序质量值小于20的碱基,小于25bp的测序读长将被丢弃,过滤采用自写Perl程序执行;
其次,用STAR软件将高质量转录组测序序列数据比对到参考序列上并进行过滤,保留只有唯一比对位置且错配数小于2%的序列比对结果,比对结果使用Samtools软件挖掘可能的变异位点,再用自写Perl程序仅保留比对质量大于phred值15、变异质量大于phred值30、只有2种基因型、总深度大于6小于100000、参考序列基因型深度大于3、变异基因型深度大于3、参考序列基因型深度比例大于5%和变异基因型深度比例大于5%的比对结果;
C、与目的基因紧密连锁的转录本的筛选:
混池筛选和目的基因紧密连锁转录本的原理是:和目的基因越近的转录本在两混池间的等位基因频率差异越大,从而通过计算转录本SNP等位基因频率差异大小可以判断其与目的基因的远近;
用自写Perl脚本从比对结果中得到SNP位点不同基因型在混池中的表达深度,以此计算等位基因频率;
另外用自写Perl脚本计算各转录本各SNP位点最可能的两基因型在高值池和低值池的等位基因频率并计算其差值,同时用Fish精确检验计算两基因型在两混池中的表达量列联表差异p-value,排除两混池间等位基因频率差值小于0.6和Fish精确检验p-value值大于1e-8的SNP位点,然后排除含有两混池间等位基因频率差值小于0.6或Fish精确检验p-value值大于1e-8的SNP位点的转录本,最后剩下的转录本我们认为是和目的基因紧密连锁的转录本;
D、分子标记开发和定位:
首先,依据得到的SNP位点设计CAPS或dCAPS标记,并依据与IWGSC数据库比对的结果找出转录本中在A/B/D同源基因间存在差异的特定位置,根据该位置设计EST标记,此外依据转录本序列和比对上的IWGSC序列设计SSR标记;
其次,在作图群体中对分子标记进行多态性检验和分型;
最后,依据表型和各标记基因型数据进行遗传定位。
由上述本发明提供的技术方案可以看出,本发明实施例提供的小麦BSR-Seq基因定位的方法,不依赖于参考基因组序列、低成本、快速、精度高。
附图说明
图1为本发明实施例中小麦抗白粉病基因PmTm4混池转录组测序高质量变异分布;
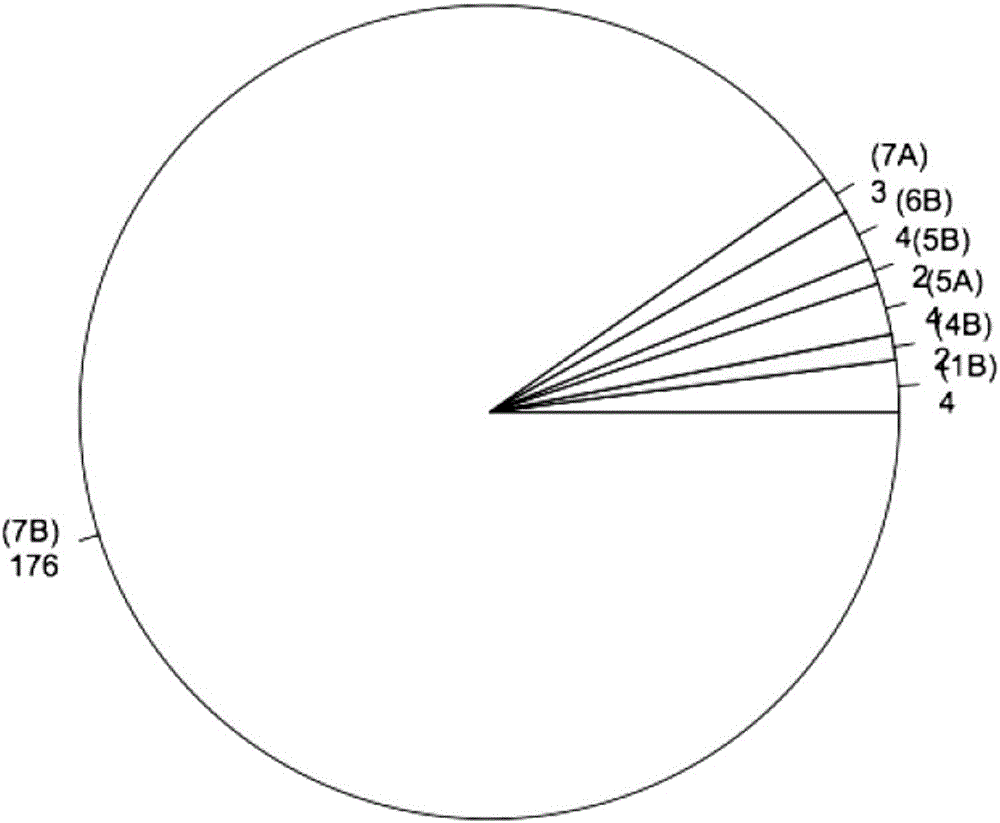
图2为本发明实施例中小麦抗白粉病基因PmTm4混池转录组测序候选SNP组成饼图;
图3为本发明实施例中小麦抗白粉病基因PmTm4的遗传图谱。
图3中染色体左边数字显示遗传图谱上标记的相对位置,染色体右边表示标记名称,和PmTm4最近的非共分离标记只存在有一个交换,Xwggc开头的标记为混池转录组测序数据分析而来的分子标记。
具体实施方式
下面将对本发明实施例作进一步地详细描述。
本发明的小麦BSR-Seq基因定位的方法,其较佳的具体实施方式是:
包括步骤:
A、混池的构建和测序:
根据重组自交系作图群体、加倍双单倍体(DH)群体、回交渗入系群体、F2或F2:3分离群体表型鉴定结果,分别用15-30个以上纯合极端高值个体和15-30以上个纯合极端低值个体分别组建高值混合池和低值混合池,在表型未表现出差异,或表现出差异后分别取等量叶片组织混合而成高值池和低值池,并提取高值池和低值池的mRNA后进行转录组测序,从而得到两个混池的转录组测序数据。
B、高质量变异挖掘:
首先,对转录组测序原始数据进行过滤得到高质量数据,过滤标准是去除两端测序质量值小于20的碱基,小于25bp的测序读长将被丢弃,过滤采用自写Perl程序执行;
其次,用STAR软件将高质量转录组测序序列数据比对到参考序列上并进行过滤,保留只有唯一比对位置且错配数小于2%的序列比对结果,比对结果使用Samtools软件挖掘可能的变异位点,再用自写Perl程序仅保留比对质量大于phred值15、变异质量大于phred值30、只有2种基因型、总深度大于6小于100000、参考序列基因型深度大于3、变异基因型深度大于3、参考序列基因型深度比例大于5%和变异基因型深度比例大于5%的比对结果;
C、与目的基因紧密连锁的转录本的筛选:
混池筛选和目的基因紧密连锁转录本的原理是:和目的基因越近的转录本在两混池间的等位基因频率差异越大,从而通过计算转录本SNP等位基因频率差异大小可以判断其与目的基因的远近;
用自写Perl脚本从比对结果中得到SNP位点不同基因型在混池中的表达深度,以此计算等位基因频率;
另外用自写Perl脚本计算各转录本各SNP位点最可能的两基因型在高值池和低值池的等位基因频率并计算其差值,同时用Fish精确检验计算两基因型在两混池中的表达量列联表差异p-value,排除两混池间等位基因频率差值小于0.6和Fish精确检验p-value值大于1e-8的SNP位点,然后排除含有两混池间等位基因频率差值小于0.6或Fish精确检验p-value值大于1e-8的SNP位点的转录本,最后剩下的转录本我们认为是和目的基因紧密连锁的转录本;
D、分子标记开发和定位:
首先,依据得到的SNP位点设计CAPS或dCAPS标记,并依据与IWGSC数据库比对的结果找出转录本中在A/B/D同源基因间存在差异的特定位置,根据该位置设计EST标记,此外依据转录本序列和比对上的IWGSC序列设计SSR标记;
其次,在作图群体中对分子标记进行多态性检验和分型;
最后,依据表型和各标记基因型数据进行遗传定位。
本发明的小麦BSR-Seq基因定位的方法,不依赖于参考基因组序列、低成本、快速、精度高。
本发明将下一代转录组测序技术(转录组测序,RNA-Seq)和混池技术(Bulked Segregant Analysis,BSA)相结合解决相关问题。首先,利用小麦测序草图序列(International Wheat Genome Sequencing Consortium 2014)作为参考序列,虽然其基因组覆盖度约60%但其基因覆盖度可达到90%,解决了小麦无完整参考转录本序列可用问题。其次,采用下一代测序技术高通量挖掘转录本上的大量的高质量SNP遗传变异,再结合混池技术精确计算等位基因频率来快速的筛选出可能与目的性状紧密连锁的转录本,并通过Fish精确检验控制假阳性。这极大的提升了小麦基因定位的效率和精度并极大的降低了小麦多态性分子标记开发的成本,使小麦基因的精细定位工作时长从数年降低到数月、定位精度从数cM降低到零点几或0cM以及精细定位成本从数万降低到数千。本发明对不同小麦性状相关基因的精细定位和克隆具有重要意义。
具体步骤:
第一,根据重组自交系作图群体、加倍双单倍体(DH)群体、回交渗入系群体、F2或F2:3分离群体表型鉴定结果,分别用15-30个以上纯合极端高值个体和15-30以上个纯合极端低值个体分别组建高值混合池和低值混合池,在表型未表现出差异,或表现出差异后分别取等量叶片组织混合而成高值池和低值池,并提取高值池和低值池的mRNA后进行转录组测序,从而得到两个混池的转录组测序数据。
第二,高质量变异挖掘。为了挖掘高质量变异,首先对转录组测序原始数据进行过滤得到高质量数据,过滤标准是去除两端测序质量值小于20的碱基,小于25bp的测序读长将被丢弃,过滤采用自写Perl程序执行;其次,用软件STAR(Dobin等,2013)将高质量转录组测序序列数据比对到参考序列上并进行过滤,保留只有唯一比对位置且错配数小于2%的序列比对结果。比对结果使用软件Samtools(Li等,2009)挖掘可能的变异位点,再用自写Perl程序仅保留比对质量大于phred值15、变异质量大于phred值30、只有2种基因型、总深度大于6小于100000、参考序列基因型深度大于3、变异基因型深度大于3、参考序列基因型深度比例大于5%和变异基因型深度比例大于5%的比对结果。
第三,与目的基因紧密连锁的转录本的筛选。混池筛选和目的基因紧密连锁转录本的原理是,和目的基因越近的转录本在两混池间的等位基因频率差异越大,从而通过计算转录本SNP等位基因频率差异大小可以判断其与目的基因的远近。用自写Perl脚本从比对结果中得到SNP位点不同基因型在混池中的表达深度,以此计算等位基因频率。另外用自写Perl脚本计算各转录本各SNP位点最可能的两基因型在高值池和低值池的等位基因频率并计算其差值,同时用Fish精确检验计算两基因型在两混池中的表达量列联表差异p-value,排除两混池间等位基因频率差值小于0.6和Fish精确检验p-value值大于1e-8的SNP位点,然后排除含有两混池间等位基因频率差值小于0.6或Fish精确检验p-value值大于1e-8的SNP位点的转录本,最后剩下的转录本我们认为是和目的基因紧密连锁的转录本。
第四,分子标记开发和定位。首先依据得到的SNP位点设计CAPS或dCAPS标记,并依据与IWGSC数据库比对的结果找出转录本中在A/B/D同源基因间存在差异的特定位置,根据该位置设计EST标记,此外依据转录本序列和比对上的IWGSC序列设计SSR标记。其次在作图群体中对分子标记进行多态性检验和分型。最后依据表型和各标记基因型数据进行遗传定位。
通过这些步骤,我们成功利用小麦测序草图作为参考转录本序列,并采用严格过滤步骤一定程度解决了小麦基因多拷贝带来的变异挖掘假阳性问题,得到了高质量变异;再利用混池原理,通过等位基因频率差异来判断连锁的转录本或变异,并结合Fish精确检验有效排除连锁假阳性的转录本。从而得到了有效的、低成本、快速和高定位精度的小麦混池转录组测序基因定位技术。
附表:
表1小麦抗白粉病基因PmTm4混池转录组测序数据的质量控制结果统计
表2小麦抗白粉病基因PmTm4混池转录组测序高质量数据比对结果统计
具体实施例:
实施例一:小麦抗白粉病基因PmTm4的混池转录组测序数据分析和精细定位
具体方法为:
(1)混池的构建和测序。为了对小麦抗白粉病基因PmTm4进行精细定,以抗病亲本唐麦4号和感病亲本农大015为亲本组合构建了包含1,504个个体的F2分离群体,并获得其F2:3家系,表型鉴定后60个纯合抗病F2:3家系和60个纯合感病F2:3家系在侵染白粉菌后3叶期对每个家系进行取样,每个家系中的一个个体的第3片叶顶端5厘米(cm)的叶片组织被采集后进行混合,抗病家系混合成抗病混池,感病家系混合成感病混池,并对混池进行RNA提取和转录组双末端测序。抗病混池的数据量为100bp长的73,229,327对Read,感病混池的数据量为100bp长的90,218,629对Read。
(2)高质量变异挖掘。通过质量控制,抗病池和感病池转录组测序数据两条双末端Read都保留下来的比例超过99%,显示测序数据质量很高,每个样本过滤后的数据总量在15Gb左右(表1)。序列比对和过滤后发现,能比对到参考序列上的Read对数大于90%,这说明参考转录本序列的完整性较高;比对到唯一位置的可信比对Read对数占比近70%,比对到多个位置的Read对数占比在23%左右,因序列差异较大无法比对到参考序列上的Read对数占比0.39%,这显示测序数据和参考序列较为相似,且小麦基因组中存在大量高度相似的基因并在过滤中被排除。另外,SNP和Indel发生的概率小于0.15%和0.02%,进一步说明了编码区测序数据和参考基因组序列的相似性;发现超过2千6百万个剪切位点,表明小麦基因组基因数可能超过预期,特别是蛋白编码基因,也说明小麦基因的剪切变体非常丰富(表2)。在抗感池转录组数据间找到SNP 256,247个,高质量SNP 106,487个,高质量SNP在各染色体上的数量和各染色体大小成正比(图1)。
(3)与目的基因紧密连锁的转录本的筛选。计算等位基因频率差异(AFD)和Fish精确检验后,发现关联的SNP位点主要位于7BL染色体臂上,这和以前的定位结果一致(Hu等,2008)。经过筛选(AFD>0.8,P-value<1e-10),寻找到195个候选SNP,其中176(90)个位于7BL上(图2),这些SNP集中在对应于短柄草1Mb区域内,这表示筛选效果很好且PmTm4很可能位于该区域内。
(4)分子标记开发和定位。选取15个候选SNP进行分子标记开发,其中11个具有预期的多态性,显示挖掘出的变异质量较高。对193个F2:3家系的重组个体进行分型,最终PmTm4基因被定为在一个0.51cM的区间内,对应1.9Mb的区间,最近的上下游非共分离标记都只存在一个交换,极大的改善了该基因的定位精度(图3)。这些结果表明通过对混池转录组数据进行分析寻找到的候选SNP和目的基因紧密连锁。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!