一种用于高效氧还原反应的Fe/N/C共掺杂电催化剂制备方法及其应用与流程
本发明属于电催化技术领域,具体涉及一种用于高效氧还原反应的fe/n/c共掺杂催化剂制备方法及其应用。
背景技术:
燃料电池作为一种新的绿色能源方式,由于其具有高效、环保等特点越来越受到人们的普遍关注。发生在燃料电池阴极的氧气还原过程(orr)是一个十分重要的半反应,但其反应动力学缓慢极大地限制了燃料电池的效率。目前,铂及其合金催化剂被认为是性能最优的orr催化剂,但其价格昂贵、储量低等因素对燃料电池的大规模商业化应用造成了巨大的局限性。因此,设计开发具有优异性能的新型廉价非贵金属催化剂来代替铂基催化剂就显得十分有必要。
fe、co、ni及其化合物等非贵金属在自然界中储量丰富,价格低廉且易得,近年来研究表明非贵金属催化剂展现出可以媲美贵金属的优异orr性能,成为构建orr燃料电池阴极催化剂的重要组成部分。其中,在非贵金属催化剂中引入一种或多种异质元素(n、s、b、p等)可以有效提升其催化活性及稳定性,性能的大幅提升可能是来源于它们独特的电子结构和通过调控形貌暴露出来的更多的活性位点,使其更容易催化氧气的还原,降低反应能垒。由于碳纳米材料(碳黑、碳纳米管、碳纳米纤维、石墨烯及纳米多孔结构碳等)具有优异的导电性和大的比表面积,因此在制备过程中通常会加入适量的碳材料做为骨架和导电基底。但大多数报道的制备方法较为复杂,在此背景下开发一种制备过程简单且性能优异的非贵金属orr催化剂就尤为必要。
本发明通过水热和高温热解的方式成功制备了在碱性条件下具有优异催化活性、稳定性和抗甲醇性能的fe/n/c共掺杂催化剂,该制备方法简单方便,原料易得。
技术实现要素:
为解决上述问题,本发明的目的之一是提供一种fe/n/c共掺杂电催化剂制备方法,该方法制备过程简单、原料易得且廉价,具备实际应用潜力。
本发明的目的之二是提供该fe/n/c共掺杂电催化剂的应用,即用于燃料电池的阴极氧还原过程,其相对于可逆氢电极的orr起始电位为0.963v,半波电位为0.821v,经耐久性测试后半波电势仅降低7mv,同时具备优异的抗甲醇性能。
为达到以上目的,本发明采取的具体的技术方案为:
1.一种用于高效氧还原反应的fe/n/c共掺杂电催化剂的制备方法,包含以下步骤:
(1)将1.0g碳黑(xc-72,比表面积为235m2g-1)放入6mol/l硝酸溶液中,水浴加热,在80℃下回流48h,然后抽滤,二次水洗涤三次,放在真空干燥箱中,60℃烘干,充分研磨置于干燥处备用。
(2)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(3)取上述黑色粉末样品(中间产物)与三聚氰胺充分混合后置于管式炉中,氮气气氛下高温热解,得到黑色固体粉末。
步骤(2)所述硝酸铁的加入量为0.5~1.5mmol;
步骤(3)所述中间产物与三聚氰胺的质量比1:5~10;
步骤(3)所述氮气气氛下高温热解温度为800~1000℃,热解时间为1h,升温速率为10℃/min。
2.以上述步骤制备的一种用于高效氧还原反应的fe/n/c共掺杂电催化剂的应用,步骤如下:
在电化学工作站上进行氧还原(orr)性能测试,采用的是标准的三电极体系,涂覆有fe/n/c共掺杂电催化剂的玻碳电极为工作电极,铂片电极为对电极,甘汞电极为参比电极;0.1mol/l氢氧化钾溶液为电解液;传统五口玻璃电解池为反应装置。测试结果表明其相对于可逆氢电极的orr起始电位为0.963v,半波电位为0.821v,经耐久性测试后半波电势仅降低7mv,同时具备优异的抗甲醇性能。
所述涂覆有fe/n/c共掺杂电催化剂的玻碳电极的制备方法如下:
称取5mgfe/n/c共掺杂电催化剂粉末,加入到0.5ml异丙醇溶液中,超声分散1h,得到均匀的黑色浆液,用微量进样器抽取10μl浆液,涂敷于洁净的玻碳电极表面,然后取10μlnafion溶液(10μl0.5wt.%的nafion溶液溶解到1.0ml异丙醇中)涂敷在催化剂表面,红外灯下干燥,即得到涂覆有fe/n/c共掺杂电催化剂的玻碳电极。
本发明的有益成果:
本发明提供了一种fe/n/c共掺杂电催化剂的制备方法以及其在燃料电池氧还原中的应用,与其他氧还原催化剂相比,具有制备过程简单,价格低廉的特点,同时在与传统的商业pt/c催化剂相比,其催化性能可以与之相媲美,半波电位相对于可逆氢电极仅低了36mv,在耐久性和抗甲醇性能方面要优于pt/c催化剂。这些优异的性能得益于fe/n元素的共掺杂改变了碳纳米材料的电子结构并提升了导电性,为催化过程中电子转移传递提供了良好的通道,有利于中间产物的吸附和脱离,加快了传质速度。本发明所提供的制备方法易于实现工业化规模应用,对降低企业成本社会能耗具有一定的意义,也为设计开发非贵金属氧还原催化剂提供了新的选择。
附图说明
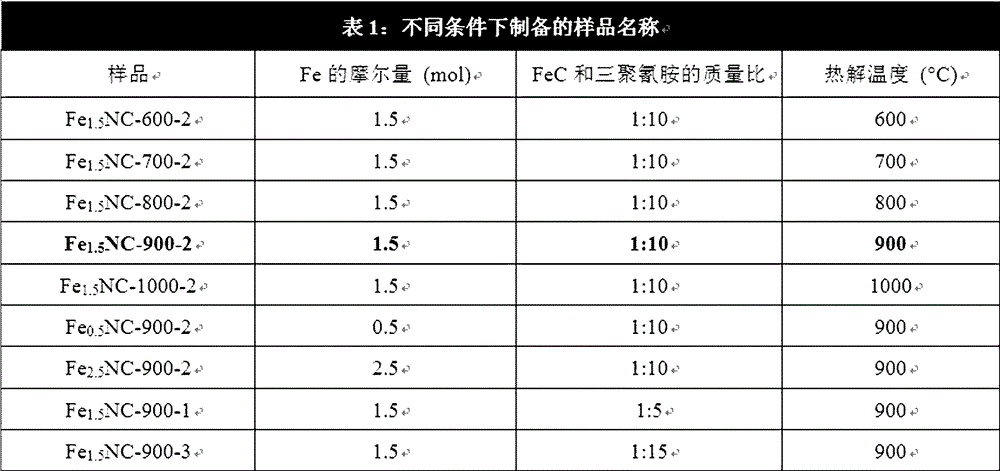
图1:不同条件下制备的样品名称。
图2:a、b为fe0.5nc-900-2的透射电镜图;c、d为fe1.5nc-900-2的透射电镜图;e、f为fe2.5nc-900-2的透射电镜图。
图3:a为fe1.5nc-600-2的透射电镜图;b为fe1.5nc-700-2的透射电镜图;c为fe1.5nc-800-2的透射电镜图;d为fe1.5nc-1000-2的透射电镜图。
图4:线(a-e)分别为fe1.5nc-t-2(t=600,700,800,900,1000)的xrd图谱,线(f-g)是fexnc-900-2(x=0.5,2.5)的xrd图谱。
图5:fe1.5nc-900-1、fe1.5nc-900-2和fe1.5nc-900-3的x射线光电子能谱,a、c和e分别为相应的总谱图;b、d、f分别为相应的n1s谱图。
图6:a为fe0.5nc-900-2、fe1.5nc-900-2和fe2.5nc-900-2的线性扫描伏安曲线;b为fe1.5nc-600-2、fe1.5nc-700-2、fe1.5nc-800-2、fe1.5nc-900-2、fe1.5nc-1000-2和pt/c的线性扫描伏安曲线;c为fe1.5nc-900-1、fe1.5nc-900-2和fe1.5nc-900-3的线性扫描伏安曲线。
图7:a为fe1.5nc-900-2经过1000圈循环伏安后的线性扫描伏安曲线对比;b为pt/c经过1000圈循环伏安后的线性扫描伏安曲线对比;c为fe1.5nc-900-2甲醇耐受性线性扫描伏安曲线对比;d为pt/c甲醇耐受性线性扫描伏安曲线对比;e为fe1.5nc-900-2和pt/c采用计时电流法的甲醇耐受性对比,f为fe1.5nc-900-2和pt/c经过1000圈循环伏安后线性扫描伏安曲线对比。
具体实施方式
以下通过具体实施例并结合附图对本发明进一步作出解释和说明。
实施例所用到的碳黑(xc-72)在使用之前,均经过以下预处理:将1.0g碳黑(xc-72,比表面积为235m2g-1)放入6mol/l硝酸溶液中,水浴加热,在80℃下回流48h,然后抽滤,二次水洗涤三次,放在真空干燥箱中,60℃烘干,充分研磨置于干燥处备用。
实施例1:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入0.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与800mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:10,氮气气氛下高温热解,热解温度为900℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe0.5nc-900-2。
实施例2:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入1.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与800mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:10,氮气气氛下高温热解,热解温度为900℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe1.5nc-900-2。
实施例3:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入2.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与800mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:10,氮气气氛下高温热解,热解温度为900℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe2.5nc-900-2。
实施例4:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入1.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与800mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:10,氮气气氛下高温热解,热解温度为600℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe1.5nc-600-2。
实施例5:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入1.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与800mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:10,氮气气氛下高温热解,热解温度为700℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe1.5nc-700-2。
实施例6:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入1.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与800mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:10,氮气气氛下高温热解,热解温度为800℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe1.5nc-800-2。
实施例7:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入1.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与800mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:10,氮气气氛下高温热解,热解温度为1000℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe1.5nc-1000-2。
实施例8:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入1.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与400mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:5,氮气气氛下高温热解,热解温度为900℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe1.5nc-900-1。
实施例9:
(1)取40mg预处理后的碳黑,1.60g葡萄糖,20mg十二烷基磺酸钠溶解到20ml二次水中,超声处理4h,形成均匀的黑色悬浮液,然后加入1.5mmol硝酸铁,将溶液转移到水热反应釜中,设置烘箱温度为180℃,反应时间为15h,自然冷却到室温,抽滤,二次水洗涤三次,真空干燥箱60℃下烘干,充分研磨后得到黑色粉末。
(2)取上80mg黑色粉末样品(中间产物)与1200mg三聚氰胺充分混合后置于管式炉中,两者的质量比为1:15,氮气气氛下高温热解,热解温度为900℃,时间1h,升温速率为10℃/min,得到黑色固体粉末,记为fe1.5nc-900-3。
如图1所示,列出了不同样品的制备条件差异以便于区分。
如图2所示,图2a中碳黑重叠度较高显示出棉绒状,观察不到明显的活性颗粒,仅有黑色团聚,由图2b可以看出原子排列十分混乱,晶格间距显示为0.209nm;随着铁盐前驱物的增加,原子排列清晰晶格间距为0.206nm,十分接近晶格间距为0.204nm的fe3n的(111)晶面;当铁盐增加到2.5mmol时,有许多不均匀的大块金属化合物形成,晶格间距为0.298nm,对应fe2o3的(220)晶面。
如图3所示,在不同热解温度下制备的催化剂样品形貌明显不同,随着热解温度的升高活性颗粒数量逐渐增加并较为均匀,但当热解温度升高到1000℃时出现不均匀的大块金属化合物。
如图4所示,xrd衍射峰在29.7°,40.8°,43.4°,57.0°处分别对应于fe3n(pdf#49-1662)的(101),(002),(111),(112)晶面和在30.2°,35.6°,57.3°,62.9°处检测到的峰分别对应于fe2o3(pdf#39-1346)的(220),(311),(422),(440)晶面,结合图2和图3所示现象表明该催化剂由碳、氧化铁和氮化铁组成。
如图5所示,图5a,5c和5e分别是是fe1.5nc-900-1,fe1.5nc-900-2和fe1.5nc-900-3的xps总谱图,进一步证明了该催化剂是由c、n、o和fe元素组成;通过比较三种催化剂的n1s图谱,可以看出在五种不同类型氮中,石墨氮和氮“铁”占40~60%,并且这两种类型的氮在fe1.5nc-900-2中的要明显高于其他两种催化剂,这可能是造成该催化剂具有优异性能的重要原因。
如图6所示,与其他催化剂相比较,fe1.5nc-900-2展现出最优的催化活性,与商业pt/c催化剂相比,半波电位仅低30mv达到了821mv(相对于可逆氢电极)。
如图7所示,图7a显示在经过1000圈的cv测试后,fe1.5nc-900-2半波电位仅降低了7mv,图7b中商业pt/c催化剂在相同的测试条件下半波电势大幅降低,达到60mv,将测试后的线性伏安曲线进行对比(图7f),可以看到fe1.5nc-900-2的半波电位要比商业pt/c高出18mv;图7c和7d分别是fe1.5nc-900-2和商业pt/c的抗甲醇性能测试,对比结果表明所制备的fe1.5nc-900-2具有优良的抗甲醇性能,为了进一步证明该催化剂的抗甲醇性能,同时采用i-t法对其进行测试(图7e),结果显示fe1.5nc-900-2在滴加甲醇后仅有微小的波动,1000s后仍维持了92%的电流,而商业pt/c大幅波动,测试结束时电流仅为54%。这些测试结果均表明fe1.5nc-900-2具有优异的orr催化性能。
- 还没有人留言评论。精彩留言会获得点赞!