一种高皮肤滞留光甘草定纳米组合物及其制备方法和用途与流程
本发明属于化妆品技术领域,具体涉及一种高皮肤滞留光甘草定纳米组合物及其制备方法和用途。
背景技术:
光甘草定是从光果甘草根中提取得到的主要黄酮类成分之一,可用作化妆品原料,被誉为“美白黄金”。光甘草定能深入皮肤内部并保持高活性,抗炎美白并高效抗氧化,有效抑制黑色素生成过程中多种酶的活性,特别是抑制酪胺酸酵素活性,同时还具有防止皮肤粗糙和抗菌的功效,被认为是疗效好、功能全面、安全性高的美白成分,是目前国际上高档美白化妆品的主要功效成分。日本、韩国化妆品公司广泛使用光甘草定制备化妆品,并且兰蔻、迪奥、香奈儿等国际品牌也在使用光甘草定,上市销售的光甘草定化妆品为霜剂、乳剂、凝胶剂或冻膜。
上述护肤功效实现的重要前提是其在皮肤组织中能够高浓度富集并长时间滞留,即实现护肤活性成分的皮肤靶向输送。但是,光甘草定在化妆品上的应用存在以下三方面的缺陷:一是光甘草定的溶解度很小、水分散性差,通常的光甘草定化妆品中活性成分质量百分含量仅为0.05~0.2%,含量低光甘草定无法达到的护肤功效;二是光甘草定容易氧化变色,降低化妆品的稳定性;三是现有上市品种通常为霜剂、乳剂,粒径较大,皮肤透过性和滞留性较差,影响护肤效果。因此,采用纳米技术和组方研究改善光甘草定的溶解性、水分散性、稳定性、皮肤透过性和滞留性,成为近年来光甘草定在美白化妆品中应用的主要研究方向和研究热点。
中国发明专利CN104367483A公开了负载光甘草定的纳米乳液及其制备方法,所制备的纳米乳液具有稳定性高、水分散性好的优点,但是所制备的纳米乳液平均粒径为130~140nm,粒径较大,光甘草定的皮肤透过性和滞留性较差。
中国发明专利CN103976892A公开了一种光甘草定微乳液及其制备方法,所制备的光甘草定微乳液包封率高,粒径大小合适,但是该技术方案的制备方法最后一步采用涡流振荡,不仅方法繁琐,而且单次制备成品总量小,不适宜工业化、规模化生产;其次,制备方法终点判断依据是溶液由浑浊变为澄清,终点评判标准无法量化。
中国发明专利CN104306352A公开了一种光甘草定微囊及其制备方法,所制备的微囊具有较好的稳定性、溶出度和流动性,且平均粒径为60~80nm,可达到缓释、控释目的,但 是本发明制备方法使用了有机溶剂二氯甲烷和石油醚,成品中会残留有毒物质二氯甲烷和石油醚,增加了光甘草定微囊使用的安全风险。
技术实现要素:
本发明的目的是提供一种高皮肤滞留光甘草定纳米组合物及其制备方法和用途,所制备的光甘草定纳米组合物不仅载药量大、稳定性好、安全性高,又可有效促进活性成分光甘草定透过皮肤角质层进入皮肤深层组织,并在皮肤中高浓度长时间滞留,且制备方法简便易控。
实现本发明的技术方案是:
一种高皮肤滞留光甘草定纳米组合物,其组分及组分质量百分比为:
一种高皮肤滞留光甘草定纳米组合物,其组分的优选质量百分比为:
一种高皮肤滞留光甘草定纳米组合物,所述固体脂质为月桂酸、肉豆蔻酸、棕榈酸、硬脂酸、山嵛酸、月桂酸甘油酯、肉豆蔻酸甘油酯、棕榈酸甘油酯、棕榈酸棕榈酯、硬脂酸甘油酯、山嵛酸甘油酯、磷脂、胆固醇、鲸蜡醇十六酸酯中的一种或几种的混合物。
一种光甘草定纳米组合物,所述液体脂质为肉豆蔻酸异丙酯、棕榈酸异丙酯、辛酸癸酸甘油三酯、亚油酸甘油酯、丙二醇单辛酸酯、维生素E、角鲨烯、大豆油中的一种或几种的混合物。
一种高皮肤滞留光甘草定纳米组合物,所述的多元醇为聚乙二醇、丙二醇、二丙二醇、 甘油、1,3-丁二醇、1,2-戊二醇中的一种或几种的混合物。
一种高皮肤滞留光甘草定纳米组合物,所述光甘草定纳米组合物的粒径为30~110nm,优选为30~80nm。
一种高皮肤滞留光甘草定纳米组合物的制备方法,所述制备方法包括以下步骤:
(1)制备油相:将2.0%~10.0%固体脂质体、1.0%~6.0%液体脂质体、0.5%~5.0%辛基十二醇在65~90℃水浴温度下熔融,熔融后再加入0.1%~6.0%光甘草定,混合均匀,备用;
(2)制备水相:将0.5%~5.0%维生素E琥珀酸聚乙二醇酯、5.0%~20.0%多元醇加入到配方量纯化水中,搅拌并水浴加热至65~90℃使维生素E琥珀酸聚乙二醇酯、多元醇溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)所制备的水相中,并不断搅拌,再5000~10000rpm高速剪切乳化1~10min,制得微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质设备或高压微射流设备处理,压力为500bar~1800bar,循环次数为2~10次,冷却至室温,即得光甘草定纳米组合物。
一种高皮肤滞留光甘草定纳米组合物在美白护肤化妆品中的应用。光甘草定纳米组合物可以直接应用于各类化妆品中,如霜剂、乳液、精华液、面膜、凝胶剂等。
皮肤角质层是表皮的最外层,由已经死亡的扁角质细胞与其间的脂质成分形成多层致密膜结构,成为人体的天然屏障。因此,护肤活性成分透皮吸收必须克服角质层的屏障作用,同时,护肤活性成分透过皮肤角质层后,还必须在护肤靶部位(活性表皮和真皮组织)中高浓度长时间滞留,才能充分发挥其护肤功效。
本发明的发明人研究发现,高皮肤滞留光甘草定纳米组合物的组分及组分比例与其促进活性成分透过皮肤角质层和提高皮肤滞留性能,以及改善纳米组合物稳定性密切相关。组合物中磷脂和胆固醇所组成的脂质体结构能够进入皮肤角质层细胞内部,与角蛋白相互作用,降低角质层细胞的致密程度,并构成脂质通道,促进光甘草定透过角质层;肉豆蔻酸异丙酯作为油相成分使用,也作为透皮促进剂,极易被皮肤吸收,辛基十二醇作为O/W型乳化剂,又具有透皮促进作用,可促进光甘草定透皮吸收,而且辛基十二醇安全性好,对皮肤无刺激,维生素E琥珀酸聚乙二醇酯是水溶性的维生素E衍生物,由亲水的极性聚乙二醇链段和亲酯的非极性维生素E琥珀酸酯链段组成,可以抑制皮肤细胞内的P-糖蛋白将护肤活性成分光甘草定外排到血液中,从而增加光甘草定在皮肤组织的滞留时间。维生素E琥珀酸聚乙二醇酯也是一类良好的O/W型乳化剂,与辛基十二醇、肉豆蔻异丙酯配合使用,可以增强纳米组合物的透皮能力,有利于活性成分光甘草定透过皮肤角质层,又可以减缓光甘草定在皮肤组织 中清除速度,延长光甘草定的滞留时间,同时能改善光甘草定溶解性和水分散性,避免其在放置过程结晶析出,提高光甘草定纳米组合物稳定性。纳米组合物中光甘草定含量高,可以发挥储库的作用,持续释放活性成分,使其较长时间维持在有效浓度,更好地发挥护肤功效。
本发明的发明人研究还发现,高皮肤滞留光甘草定纳米组合物的皮肤滞留量与其粒径密切相关,存在显著的粒径效应,当纳米组合物粒径为30~110nm时光甘草定皮肤透过和皮肤滞留效果较好,粒径为30~80nm时更佳。粒径小于30nm,纳米组合物的载药量小、滞留量小,且纳米组合物的稳定性降低;粒径大于110nm,其皮肤透过和皮肤滞留效果明显变差。
本发明的有益效果在于:
1、通过合理组方提高了光甘草定纳米组合物的皮肤透过性和滞留性:组方中磷脂和胆固醇能构成脂质通道,肉豆蔻酸异丙酯和辛基十二醇能有效促进光甘草定透皮吸收,维生素E琥珀酸聚乙二醇酯能够抑制皮肤细胞内的P-糖蛋白将护肤活性成分外排到血液中,减缓光甘草定在皮肤组织中清除速度,延长光甘草定的滞留时间。
2、本发明的光甘草定纳米组物具有合适的粒径,粒径为30~110nm,该粒径既能保证光甘草定透过角质层,又能保证光甘草定在皮肤中有较长的滞留时间,可显著提高光甘草定皮肤透过量和皮肤滞留量。
3、本发明光甘草定纳米组合物载药量大,活性成分光甘草定浓度可达6.0%,光甘草定纳米组合物透过皮肤可以作为储库,持续释放光甘草定,较长时间维持在有效浓度,更好地发挥护肤功效。
4、本发明的光甘草定纳米组合物具有良好的稳定性:一是组方中包含的维生素E琥珀酸聚乙二醇酯具有生育酚酯结构,具有抗氧化性,可避免光甘草定氧化变色,提高光甘草定的稳定性;二是组方中的维生素E琥珀酸聚乙二醇酯与辛基十二醇、肉豆蔻酸异丙酯联合使用,能提高皮肤透过量,持续释放使其较长时间维持在有效浓度,更好发挥护肤功效。
5、本发明的光甘草定纳米组合物安全性高,制备方法中未使用有机溶剂,无有机溶剂残留,对皮肤刺激小,安全性高。
6、本发明的制备方法简单,且工艺易于控制,适宜工业化、规模化生产。
附图说明
图1是实施例9制备的光甘草定纳米组合物粒径分布图。
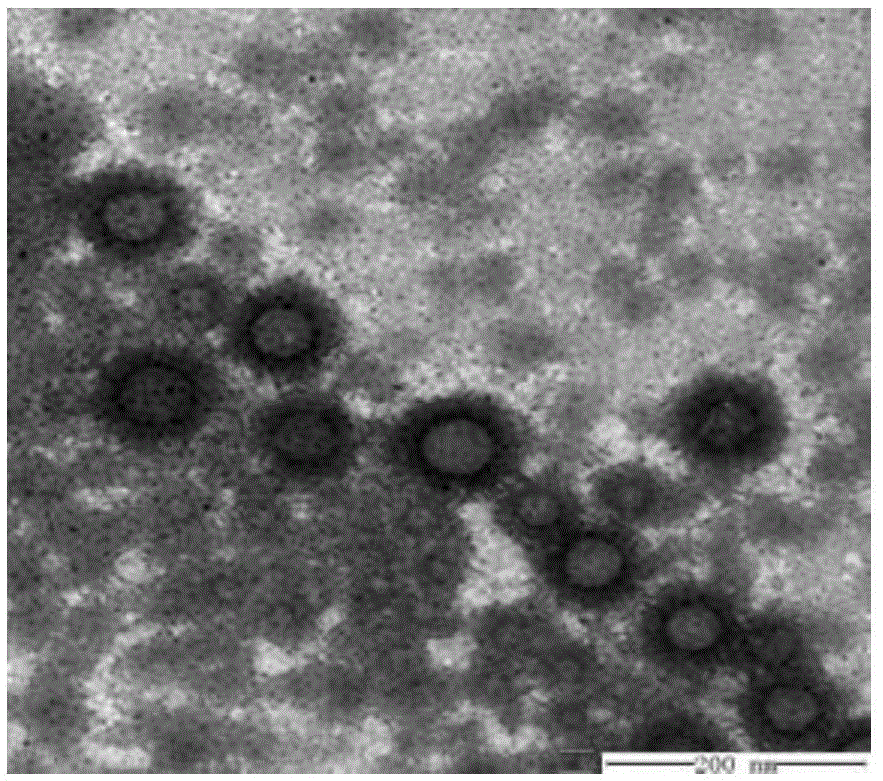
图2是实施例9制备的光甘草定纳米组合物透射电镜图。
图3是光甘草定纳米组合物体外释放曲线。
图4是光甘草定纳米组合物体外皮肤累积透过曲线。
图5是光甘草定纳米组合物体外皮肤累积透过量和滞留量比较。
图6是光甘草定纳米组合物体外皮肤累计透过量与粒径的关系。
图7是光甘草定纳米组合物体外皮肤累计滞留量与粒径的关系。
图8是小鼠使用光甘草定纳米组合物前皮肤生理切片。
图9是小鼠使用光甘草定纳米组合物后皮肤生理切片。
具体实施方式
下面以实施例方式对本发明作进一说明,使本领域技术人员可以更好的理解本发明。本发明所优选的具体实施方式应被理解为仅是举例说明,而非以任何方式限制本发明。
一、本发明实施例部分
实施例1
(1)制备油相:2.0%山嵛酸三甘油酯、1.0%辛酸癸酸甘油三酯、0.5%辛基十二醇,于65℃水浴使熔融;熔融后再加入0.1%光甘草定,混合均匀,备用;
(2)水相水相:0.5%维生素E琥珀酸聚乙二醇酯、10.0%甘油加入到85.9%纯化水中,于65℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,再5000rpm高速剪切乳化10min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质1200bar循环2次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为65.2nm,包封率为81.7%。
实施例2
(1)制备油相:4.0%大豆磷脂、1.0%角鲨烯、1.0%辛酸癸酸甘油酯、0.5%辛基十二醇,于70℃水浴使熔融;熔融后再加入0.5%光甘草定,混合均匀,备用;
(2)制备水相:1.0%维生素E琥珀酸聚乙二醇酯、5.0%1,3-丁二醇加入到87.0%纯化水中,于70℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,再8000rpm高速剪切乳化3min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压微射流1200bar循环3次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为57.4nm,包封率为78.9%。
实施例3
(1)制备油相:2.0%单硬脂酸甘油酯、2.0%月桂酸甘油酯、2.0%肉豆蔻酸异丙酯、1.0%大豆油、1.0%辛基十二醇,于90℃水浴使熔融;熔融后再加入1.0%光甘草定,混合均匀,备用;
(2)制备水相:1.5%维生素E琥珀酸聚乙二醇酯、7.0%甘油、9.0%1,2-戊二醇加入到73.5%纯化水中,于90℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,6000rpm高速剪切乳化5min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质500bar循环10次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为51.9nm,包封率为67.5%。
实施例4
(1)制备油相:4.0%大豆磷脂、5.0%棕榈酸棕榈酯、1.0%胆固醇、3.0%肉豆蔻酸异丙酯、2.0%大豆油、1.0%维生素E、3.0%辛基十二醇,于70℃水浴使熔融;熔融后再加入6.0%光甘草定,混合均匀,备用;
(2)制备水相:2.0%维生素E琥珀酸聚乙二醇酯、5.0%聚乙二醇400、10.0%丙二醇加入到58.0%纯化水中,于70℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,8000rpm高速剪切乳化8min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质1500bar循环5次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为79.5nm,包封率为88.3%。
实施例5
(1)制备油相:2.0%山嵛酸三甘油酯、3.0%棕榈酸棕榈酯、1.0%月桂酸甘油酯、1.0%肉豆蔻酸异丙酯、2.0%辛酸癸酸甘油酯、1.0%辛基十二醇,于80℃水浴使熔融;熔融后再加入3.0%光甘草定,混合均匀,备用;
(2)制备水相:5.0%维生素E琥珀酸聚乙二醇酯、8.0%1,3-丁二醇、12%丙二醇加入到62.0%纯化水中,于80℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,再8000rpm高速剪切乳化5min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质1800bar循环4次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为28.7nm,包封率为 45.6%。
实施例6
(1)制备油相:3.0%大豆磷脂、2.0%月桂酸甘油酯、1.0%肉豆蔻酸异丙酯、2.0%辛酸癸酸甘油酯、2.0%辛基十二醇,于70℃水浴使熔融;熔融后再加入3.0%光甘草定,混合均匀,备用;
(2)制备水相:1.0%维生素E琥珀酸聚乙二醇酯、10.0%甘油、10%丙二醇加入到66.0%纯化水中,于70℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,再6000rpm高速剪切乳化5min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质800bar循环4次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为67.4nm,包封率为83.4%。
实施例7
(1)制备油相:1.0%大豆磷脂、2.0%棕榈酸棕榈酯、1.0%胆固醇、1.0%肉豆蔻酸异丙酯、1.0%辛酸癸酸甘油酯、1.0%辛基十二醇,于70℃水浴使熔融;熔融后再加入2.0%光甘草定,混合均匀,备用;
(2)制备水相:1.0%维生素E琥珀酸聚乙二醇酯、10%1,3-丁二醇、10%丙二醇加入到70.0%纯化水中,于70℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,再6000rpm高速剪切乳化5min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质800bar循环4次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为70.5nm,包封率为89.7%。
实施例8
(1)制备油相:3.0%大豆磷脂、2.0%棕榈酸棕榈酯、3.0%肉豆蔻酸异丙酯、5.0%辛基十二醇,于80℃水浴使熔融;熔融后再加入2.0%光甘草定,混合均匀,备用;
(2)制备水相:1.0%维生素E琥珀酸聚乙二醇酯、15.0%1,3-丁二醇、5%丙二醇加入到64.0纯化水中,于80℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,10000rpm高速剪切乳化1min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质1500bar循环5 次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为31.5nm,包封率为52.4%。
实施例9
(1)制备油相:3.0%大豆磷脂、2.0%棕榈酸棕榈酯、4.0%肉豆蔻酸异丙酯、2.0%辛基十二醇,于70℃水浴使熔融;熔融后再加入2.0%光甘草定,混合均匀,备用;
(2)制备水相:2.0%维生素E琥珀酸聚乙二醇酯、12.0%1,3-丁二醇、8.0%丙二醇加入到65.0%纯化水中,于70℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,8000rpm高速剪切乳化5min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质1000bar循环4次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为63.7nm,包封率为87.8%。
实施例10
(1)制备油相:2.0%大豆磷脂、3.0%棕榈酸棕榈酯、3.0%肉豆蔻酸异丙酯、1.0%辛基十二醇,于70℃水浴使熔融;熔融后再加入2.0%光甘草定,混合均匀,备用;
(2)制备水相:3.0%维生素E琥珀酸聚乙二醇酯、10.0%1,3-丁二醇、5.0%丙二醇加入到71.0%纯化水中,于70℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,再6000rpm高速剪切乳化5min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质800bar循环3次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为78.5nm,包封率为85.5%。
实施例11
(1)制备油相:3.0%大豆磷脂、1.0%棕榈酸棕榈酯、2.0%肉豆蔻酸异丙酯、2.0%辛基十二醇,于70℃水浴使熔融;熔融后再加入2.0%光甘草定,混合均匀,备用;
(2)制备水相:1.0%维生素E琥珀酸聚乙二醇酯、10.0%1,3-丁二醇、5.0%丙二醇加入到74.0%纯化水中,于70℃水浴搅拌溶解,备用;
(3)制备微米级分散体:将步骤(1)所制备的油相滴加至步骤(2)制备的水相中,并不断搅拌,再6000rpm高速剪切乳化5min,制成微米级分散体;
(4)制备纳米组合物:将步骤(3)所制备的微米级分散体通过高压均质500bar循环5次,得光甘草定纳米组合物。该光甘草定纳米组合物粒径为109.8nm,包封率为 92.3%。
二、对比实施例部分
对比例1光甘草定霜剂的制备
按照常规霜剂制备方法,以1.0%光甘草定、3.0%硬脂酸、4.5%单硬脂酸甘油酯、5.0%十六十八醇、6.0%聚二甲基硅氧烷为油相,于75℃水浴中熔融;以10.0%甘油、5.0%丙二醇、0.3%三乙醇胺和65.2%纯化水作为水相,于75℃水浴中溶解;搅拌混合两相并乳化,冷却,即得含量为1.0%的光甘草定霜剂,作为实施例8、实施例9、实施例10和实施例11的对照制剂。
对比例2纳米光甘草定组合物霜剂的制备
按对比例1中的配方及方法制备的空白霜剂与实施例9样品1:1复配,所得的霜剂约含1%的光甘草定,作为霜剂对照。
三、试验例部分
实验例1光甘草定纳米组合物的稳定性对比分析
实施例1~11光甘草定纳米组合物在密闭容器在室温条件下放置30天后,检查样品的性状及粒径。
表1 实施例1~11光甘草定纳米组合物稳定性结果
试验结果表明:本发明所制备的实施例1~11样品无团聚现象,且粒径在30~110nm之间,满足实际应用要求,所制备的样品放置30天后,性状及粒径未发生显著性变化,仍然满 足实际应用需求,表明按照本发明所制备光甘草定纳米组合物性状稳定,尤其在药物浓度高的情况下仍然较稳定。未发现光甘草定结晶析出现象,无药物泄漏。
实验例2光甘草定体外释放比较
分别取实施例8~11、对比例1、对比例2样品各1g于透析袋内,将透析袋置于装有50mL 2%Tween 80-20%乙醇-生理盐水释放介质的锥形瓶中,于电热恒温调速振荡器中(37±0.5)℃、100rpm振摇释放,并于1h,2h,4h,6h,8h,10h,24h分别取样1mL,再迅速补充1mL相同温度下的空白释放介质。样品用0.22μm有机滤膜过滤后,HPLC进样检测,计算药物不同时间的累积释放度。
光甘草定纳米组合物及对比例2的释放度均高于对比例1,粒径较小的实施例8相对于其他实施例9、10、11相对较快,并且对比例2的释放行为与光甘草定纳米组合物基本一致。
实验例3光甘草定纳米组合物的皮肤透过性和滞留性的对比分析
以体重为200~250g雄性SD大鼠腹部皮肤为透皮试验的障碍层。将完整无破损的皮肤固定于接收池和供给池之间(皮肤内层面向接收池)。扩散池参数:有效扩散面积3.14cm2,接收池容积约8.0ml,磁力搅拌速度600rpm。在接收池内充满释放介质2%Tween 80-20%乙醇-生理盐水,排除气泡,开启搅拌,并恒温至(37.0±0.5)℃。向皮肤表面分别均匀涂布样品约1g(n=8),于1h、2h、4h、6h、8h、10h、12h、24h吸取接收液0.5ml,并补充释放介质0.5ml。用HPLC测定经0.22μm滤膜过滤的接收液中光甘草定的浓度。计算不同时间药物累积透过量。
按以下公式计算光甘草定单位面积累积透皮量:
其中,Q:累积透皮量;S:有效扩散面积;V:接收池中生理盐水体积;Ci:第1次至上次取样时接收液中药物浓度;Cn:该次取样时接收液中药物浓度;m0:样品理论称样量;m:样品实际称样量。
将本发明实施例8~11样品、对比例1样品进行透皮试验。试验结果参见图4和图5。从图4中可以看出,实施例8~11样品的皮肤累积透过量都高于对比例1样品。从图5中可以看出本发明实施例8~11的24h累积透过量分别为1105.34μg/cm2、915.6μg/cm2、696.38μg/cm2、521.1μg/cm2,对比例1仅为81.98μg/cm2;本发明实施例8~11的24小时滞留量分别为85.5μg/cm2、191.2μg/cm2、147.0μg/cm2、72.2μg/cm2;对比例1仅为18.24μg/cm2。显然,本发明的光甘草定纳米组合物具有很高的皮肤透过量和滞留量,因而具有优 良的护肤效果。
图6和图7为本发明实施例8~11的粒径与透过量和粒径与滞留量的关系分析:从图6中可以看出,当粒径由31.5nm增大至109.8nm时,皮肤透过量由1105.34μg/cm2降低至521.1μg/cm2,显示粒径越小,透过量越大;从图7中可以看出,粒径在31.5nm至109.8nm之间时,滞留量明显较优。综合考虑粒径与皮肤透过量和滞留量的关系,本发明选择的粒径范围在30~80nm之间,能够较好的兼顾皮肤透过量和皮肤滞留量,以起到更好的皮肤屏障修护、保湿等护肤效果。
实验例4皮肤功效评价
小鼠皮肤每天涂抹对比例2(对比例1的空白霜剂与实施例9样品1:1复配),1次/天,30天后小鼠皮肤组织一般形态学如图8、图9所示。
涂抹光甘草定纳米组合物的小鼠皮肤结构完整,层次较清晰,表皮基底层与真皮层结构清晰真皮层纤维组织排列排列整齐,呈现波浪状,细胞分层较清晰,细胞间质分布均匀。与比涂抹前比较,小鼠皮肤组织一般形态有明显改善。
实验例5皮肤刺激性评价
①试验设计
取健康家兔18只,体重(2.0±0.2)kg,随机分为6组,每组动物3只,于实验前24h将家兔背部皮肤两侧去毛,去毛后24h检查去毛皮肤是否受伤,受伤皮肤不宜做皮肤刺激性试验。每天涂抹3次,连续涂抹7天。包括空白霜剂组(不给予任何药物)、比较实施例8~11分别与对比例1中空白霜剂1:1复配进行皮肤刺激性试验。
②试验结果见表2
表2 实施例8~11及空白组皮肤刺激性观察结果
“+”家兔皮肤充血、红肿;“++”表示充血、红肿现象仍在,但有增加趋势;“—”表示无充血、红肿现象。
试验结果表明:本发明实施例8~11分别与对比例1空白霜剂1:1复配的样品涂抹于家 兔皮肤无充血、红肿现象,表明实施例8~11与霜剂均无刺激性。
- 还没有人留言评论。精彩留言会获得点赞!