用于乙基纤维素聚合物分散体的分批混合方法与流程
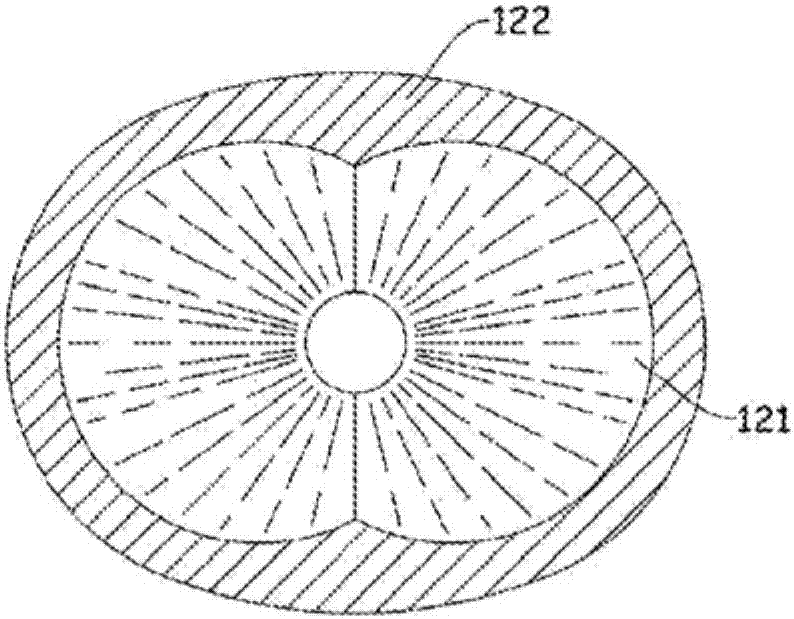
通常需要制备含有乙基纤维素聚合物的膜。此膜例如可用作施加到其它膜或珠粒的涂层。在一些情况下,珠粒的集合体包含药物,并且然后利用含有乙基纤维素聚合物的膜包覆那些珠粒中的每一个。当将珠粒放在诸如可在人体中发现的含水环境中时,含有乙基纤维素聚合物的膜可以提供药物的控释。也希望膜具有良好的机械性能,例如高拉伸强度、高拉伸伸长率、和/或表面平滑度。在过去,制备这种膜的常用方法是将珠粒与其中乙基纤维素聚合物溶解在有机溶剂中的溶液接触。由于环境和健康的影响,有机溶剂是不合需要的。希望提供含有乙基纤维素聚合物并且能够制备高质量膜的含水涂料组合物。这种含水涂料组合物的一种理想形式是含水乙基纤维素聚合物分散体,其是乙基纤维素聚合物颗粒分散在连续含水介质中的形式。用于制备这种含水组合物的一些先前已知的步骤是连续方法,例如使用挤出机的方法。连续方法通常苦于以下缺点中的一个或多个:连续方法通常不太适合制备相对小体积的材料,并且连续方法通常难以对组合物的组分进行频繁的改变。us4,502,888描述了制备使用脂肪酸盐作为增塑剂/稳定剂的水不溶性聚合物的分散体的方法。期望提供一种避免连续方法的缺点中的一个或多个的分批方法。也希望提供能够制备具有理想的小粒径的含水乙基纤维素聚合物分散体的分批方法。以下是本发明的陈述。本发明的第一方面是一种制备含水组合物的方法,其包含(a)提供包含可密封容积和在所述可密封容积内包含一个或多个转子的混合器;(b)将包含乙基纤维素聚合物和脂肪酸的成分放入所述可密封容积中;(c)将包含水和水溶性碱的成分放入所述可密封容积中;(d)在所述步骤(b)和(c)后密封所述可密封容积;(e)然后旋转所述转子中的一个或多个,同时使所述成分处于高于所述乙基纤维素聚合物的软化点的温度下,以产生所述含水组合物;其中进行所述步骤(e),使得所述成分的体积的90%或更少不与所述转子中的一个或多个接触;其中所述含水组合物包含分散在含水介质中的颗粒,其中所述颗粒包含部分或全部所述乙基纤维素聚合物,并且其中所述含水介质包含部分或全部所述水。以下是附图的简要说明。图1是用于本发明的一个实施例的混合器的垂直截面。图2是图1中所示的混合器的下壳体的水平截面。图3也是图1中所示的混合器的下壳体的水平截面。以下是本发明的具体实施方式。如本文所用,以下术语具有指定的定义,除非上下文另有明确指出。如本文所用,含水组合物具有基于组合物的重量的20重量%或更多的水。如本文所用,分散体是包含在25℃下呈液体的连续介质并且包含分布在整个连续液体介质中的物质的离散颗粒(本文称为“分散颗粒”)的组合物。如本文所用,含水分散体是一种含水组合物,其是连续液体介质包含基于连续液体介质的重量的50重量%或更多的水的分散体。溶解在连续液体介质中的物质在本文被认为是连续液体介质的一部分。所有分散颗粒的集合体在本文中被称为分散体的“固相”。如本文所用,含水组合物的“固体含量”是当除去了水和具有250℃或更低的沸点的化合物时残留的材料的量。固体含量的特征在于基于含水组合物的总重量的重量百分比或基于含水组合物的总体积的体积分数。如本文所用,乙基纤维素聚合物意指纤维素的衍生物,其中葡萄糖重复单元上的一些羟基被转化为乙醚基。乙醚基的数量可以变化。乙醚含量的usp专论要求是44%至51%。如本文所用,乙基纤维素聚合物的粘度是基于溶液重量的该乙基纤维素聚合物于溶剂中的5重量%溶液的粘度。溶剂是80重量%甲苯和20重量%乙醇的混合物。在乌氏(ubbelohde)粘度计中在25℃下测量溶液的粘度。如本文所用,脂肪酸是具有羧基和脂肪基的化合物。脂肪基是含有8个或更多个碳原子的彼此连接的碳原子的直链或支链。烃脂肪基仅含有碳原子和氢原子。如本文所用,增塑剂是可与乙基纤维素聚合物混溶并且当与乙基纤维素聚合物混合时其降低该乙基纤维素聚合物的玻璃化转变温度的化合物。如果在25℃下2克或更多的化合物溶解于100克水中,那么本文认为化合物是水溶性的。即使需要将水加热到高于25℃的温度以形成溶液,只要在25℃下2克或更多的化合物在水中的溶液是稳定溶液,那么化合物被认为是水溶性的。如本文所用,“聚合物”是由较小化学重复单元的反应产物组成的相对较大的分子。聚合物可以具有单一类型的重复单元(“均聚物”),或它们可以具有多于一种类型的重复单元(“共聚物”)。共聚物可以具有随机、顺序、嵌段、以其它布置形式布置的各种类型的重复单元,或其任何混合物或组合。聚合物具有2,000道尔顿或更高的重均分子量。材料的软化点是一温度,低于所述温度时材料表现为固体并且高于所述温度时材料在轻度至中度的应力下开始能够流动。软化点根据astme28-14通过环球法测量。如本文所用,碱是一种具有接受质子以形成化合物的共轭酸的能力的化合物,并且该化合物的共轭酸具有7.5或更大的pka。如本文所用,脂肪酸是具有羧基和脂肪基的化合物。脂肪基是含有8个或更多个碳原子的彼此连接的碳原子的直链或支链。烃脂肪基仅含有碳原子和氢原子。如本文所用,“多颗粒”是多个颗粒。颗粒在25℃下是固体。颗粒是球形或接近球形。如果颗粒不是球形,则其直径在文中被认为是具有相同体积的球体的直径。如果容器可以打开以允许将成分放入容器中然后密封,使得如果容器内的成分达到0.55mpa(80psig)或更少的压力,容器将不会泄露,则容器在文中被视为是“可密封的”。当本文指出比率是x:1或更大时,这意指该比率是y:1,其中y等于或大于x。例如,如果指出某个比率是0.2:1或更大,该比率可以是0.2:1或0.5:1或100:1,但该比率不是0.1:1或0.02:1。类似地,当本文指出比率是w:1或更小时,这意指该比率是z:1,其中z等于或小于w。例如,如果指出某个比率是5:1或更小,该比率可以是5:1或4:1或0.1:1,但该比率不是6:1或10:1。在本发明中可以使用任何乙基纤维素聚合物。乙基纤维素聚合物的乙醚含量是44%或更高;优选地47%或更高;更优选地48%或更高。乙基纤维素聚合物的乙醚含量是51%或更低;优选地50%或更低。乙基纤维素聚合物优选地具有2mpa-s或更高;更优选地5mpa-s或更高;更优选地12mpa-s或更高;更优选地16mpa-s或更高的粘度。乙基纤维素聚合物优选地具有120mpa-s或更低;更优选地100mpa-s或更低;更优选地80mpa-s或更低;更优选地60mpa-s或更低;更优选地40mpa-s或更低;更优选地30mpa-s或更低的粘度。乙基纤维素优选地具有120℃或更高;更优选地130℃或更高的软化点。乙基纤维素优选地具有160℃或更低;更优选地150℃或更低;更优选地140℃或更低的软化点。可用于本发明的乙基纤维素聚合物的市售形式包括例如可从陶氏化学公司(thedowchemicalcompany)以商标ethoceltm购得的那些。用于本发明实例的乙基纤维素聚合物可从陶氏化学公司(thedowchemicalcompany)以具有48.0%至49.5%的乙醚含量的ethoceltm标准4、ethoceltm标准7、ethoceltm标准10、ethoceltm标准20、ethoceltm标准45、或ethoceltm标准100购得。可用于本发明实施例的其它市售的乙基纤维素聚合物包括可购自ashland,inc.的某些等级的aqualontm乙基纤维素和可购自ashacellulosepvt.ltd的某些等级的ashaceltm乙基纤维素聚合物。本发明涉及含水分散体。优选地,连续液体介质包含基于连续液体介质的重量的60重量%或更多;更优选地70重量%或更多;更优选地80重量%或更多;更优选地90重量%或更多的量的水。优选地,含水分散体中的分散颗粒含有基于固相的总干重的40重量%或更多;更优选地50重量%或更多;更优选地60重量%或更多的量的乙基纤维素聚合物。优选地,含水分散体中的分散颗粒含有基于固相的总干重的90重量%或更少;更优选地80重量%或更少的量的乙基纤维素聚合物。分散颗粒在文中被认为包含位于颗粒内部的材料和位于颗粒表面上的材料,比如,例如,分散剂。本发明的组合物含有一种或多种脂肪酸,其可以是饱和的或不饱和的。更优选地是不饱和脂肪酸。脂肪酸的脂肪基可以是直链或支链的;优选地是直链的。脂肪酸的脂肪基可以是烃脂肪基或可以具有除氢或碳外的一种或多种取代基,优选地是烃脂肪基。在不饱和脂肪酸中,优选地是肉豆蔻烯酸、棕榈油酸、棕榈酸(sapienicacid)、油酸、亚油酸、和花生四烯酸。在饱和脂肪酸中,优选地是辛酸、癸酸、月桂酸、棕榈酸、肉豆蔻酸、硬脂酸、和花生酸。最优选地是油酸。优选地,脂肪酸的量是基于固相的总干重的2重量%或更多;更优选地4重量%或更多;更优选地6重量%或更多。优选地,脂肪酸的量是基于固相的总干重的20重量%或更少;更优选地18重量%或更少;更优选地12重量%或更少。本发明的组合物含有一种或多种碱。碱是水溶性的。优选的碱是氨、有机胺、碱金属氢氧化物、和碱土金属氢氧化物。更优选的碱是氨和碱金属氢氧化物。最优选的碱是碱金属氢氧化物。稳定性碱优于不稳定性碱。在不稳定性碱中,优选地是氨和作为有机胺的不稳定性碱。在不稳定性碱中,更优选地是氨。在有机胺不稳定性碱中,优选地是吗啉、醇胺、及其混合物;更优选地是氨、吗啉、二乙醇胺、2-氨基-2-甲基-1-丙醇、及其混合物。优选地,碱与脂肪酸的碱:酸当量比是1:1或更高;更优选地是1.1:1或更高。优选地,碱与脂肪酸的碱:酸当量比是10:1或更低。当碱由一种或多种稳定性碱组成时,优选地,碱化合物与脂肪酸的碱:酸当量比是2:1或更低;更优选地是1.5:1或更低。当碱由一种或多种不稳定性碱组成时,优选地,碱与脂肪酸的碱:酸当量比是5:1或更高;更优选地是6:1或更高;更优选地是7:1或更高。当碱由一种或多种不稳定性碱与一种或多种稳定性碱的混合物组成时,优选地,所有不稳定性碱的聚集体与脂肪酸的碱:酸当量比按照上述针对碱由一种或多种不稳定性碱组成的情况中的碱:酸当量比所述的优选值,并且优选地是,所有稳定性碱的聚集体与脂肪酸的碱:酸当量比按照上述针对碱由一种或多种稳定性碱组成的情况中的碱:酸当量比所述的优选值。本发明的方法涉及混合器的使用。混合器是包括可密封容积的装置。也就是说,装置的一部分是可以放置各种成分的容器,并且容器可以被密封。可密封容积的大小优选地是10ml或更大;更优选地是50ml或更大。可密封容积的大小优选地是1,000l或更小。混合器也包括一个或多个转子。转子是围绕轴旋转的机械元件。转子在可密封容积内旋转。优选地,混合器包括两个或更多个转子。优选地,每个转子由穿过混合器的壳体的驱动轴驱动。优选地,混合器的壳体具有驱动轴可以穿过的端口;优选地,端口允许驱动轴由位于混合器外部的电动机驱动,同时保持密封,其当可密封容积内的成分处于升高的压力时防止泄漏。转子可以具有任何形状或设计。一些合适的转子是圆柱形或锥形的并且围绕圆柱体或锥体的轴线旋转。在圆柱形或锥形转子中,优选地是在表面上具有一个或多个螺旋槽的那些。圆柱形或锥形转子的实例是用于挤出机的螺杆,单螺杆或双螺杆,以及用于研究聚合物熔体行为而设计的内部混合器的桨叶。转子的优选形状是圆柱形或锥形螺旋中的带状。也就是说,带是符合假想圆柱体或假想锥体的表面的螺旋形状。转子围绕假想圆柱体或锥体的轴旋转。优选地是锥形螺旋。更优选地是具有相互啮合并沿相反方向旋转的两个相对的锥形螺旋转子的混合器。也希望是固定到围绕轴的轴线旋转的轴的一个或多个叶轮的形状的转子。使用叶轮形状的转子的混合器的实例由parr仪器公司制造。当材料在混合器中并且转子旋转时,用于表征由一个或多个所述转子“接触”的材料的体积。对于该表征,“材料”指存在于混合器中的液体和固体化合物或混合物。在步骤(e)期间选择时刻(“分析时刻”),并且考虑材料所占据的空间(标记为sm0)。sm0是材料所占据的空间并且不包括任何转子所占据的空间。由于一些或全部转子可能被材料包围,所以sm0的形状可能非常复杂;sm0可能具有对应于转子在分析时刻存在的位置的空隙。为了表征接触体积,sm0的形状和尺寸被认为保持不变。然后,当转子旋转时,转子上的某一点与sm0中的某一点接触,并且这样的点被认为是接触的。空间sm0的体积是vm0。在转子完成了一个或多个旋转循环后,可以观察所有的接触点,并且所有这些接触点的总体积是接触体积(标记为vcon)。未接触的体积是vunc=vm0-vcon。以vm0的百分比表示的未接触的体积是%vunc=100xvunc/vm0%vunc优选地是90%或更少;更优选地80%或更少;更优选地70%或更少;更优选地60%或更少;更优选地50%或更少;更优选地40%或更少;更优选地30%或更少。优选地,当转子旋转时,一个或多个转子的某些部分靠近包含可密封容积的容器的内表面。由任何转子的任何部分到容器的内表面的最接近的距离是混合器的“间隙”。优选地,间隙是5cm或更小;更优选地2cm或更小;更优选地1cm或更小。可用于表征与转子的某一点最接近的混合器的内表面的比例。所关注的内表面是与空间sm0接触的混合器的内表面;所关注的该表面被标记为surf0。如果在转子旋转期间的任何时刻,转子的一点以两倍间隙或更小的距离接近surf0上的点,则surf0上的该点在本文被称为与转子最接近。优选地,由最接近的点构成的surf0的面积百分比是基于surf0的面积的10%或更高;更优选地20%或更高;更优选地50%或更高。优选地,混合器配备有用于加热混合器的装置。优选地,以允许热量传递到可密封容积中的材料的方式将热量施加到混合器的外部。例如,加热流体可以以允许热量通过混合器的壁传递到可密封容积中的材料的方式循环通过混合器外部的夹套。优选地,混合器配备有允许气体注入到可密封容积密封时的可密封容积中以提高密封的可密封容积内的压力的装置。优选的要注入的气体是惰性气体,包括氮气和惰性气体。更优选地是氮气。优选地,混合器配备有允许液体注入到可密封容积密封时的可密封容积中而不损失密封的装置。优选的要注入的液体是水和溶于水中的水溶性碱的溶液。图1显示了适于本发明的一个实施例的混合器的垂直截面。混合器可以分为两个部分:碗1和盖4。将材料放入碗1中,并且使碗1和盖4在密封5处在一起。一个转子具有刀片21和驱动轴31;另一个转子具有刀片22和驱动轴32。驱动轴穿过盖4而不损害混合器保持压力的能力。刀片21和22留有被成分部分或完全占据的开放体积6。驱动轴31和32通过机械地耦合到驱动轴31和32的一个或多个电动机(未显示)旋转驱动。盖4也具有允许l液体插入开放体积6中而不损害混合器保持压力的能力的装置(未显示)。盖4也具有允许气体插入开放体积6中而不损害混合器保持压力的能力的装置(未显示)。盖4也具有允许测量传送(例如,通过一根或多根导线的电信号)到混合器外部的混合器内部的条件(例如,温度和压力)而不损害混合器保持压力的能力的装置(未显示)。图2和图3是碗1的水平截面。图2显示了碗的壁面122和碗内的内表面121。图3显示了碗的壁面112和碗内空腔的内表面111。在本发明的实施中,将材料放入混合器中。材料包括乙基纤维素聚合物、脂肪酸、水、和碱。在一些初步步骤后,乙基纤维素聚合物、脂肪酸、水、和碱位于混合器的可密封容积中,使可密封容积密封。然后使材料的混合物在转子旋转的同时经受高于乙基纤维素聚合物软化点的温度。在一些实施例(“单次”实施例)中,将包括乙基纤维素聚合物、脂肪酸、水、水溶性碱、和任选的额外成分的材料放在混合器中。当将材料放在混合器中时,混合器可以在15℃至99℃的任何温度下。步骤(b)和(c)可以以任何顺序或同时进行,乙基纤维素聚合物、脂肪酸、水、水溶性碱、和任选的额外成分以任何顺序加入。在某个方便时刻,使转子与材料接触。在材料处于可密封容积中后的某个时刻,使可密封容积密封。在使可密封容积密封前或后的某个方便时刻,将材料加热到高于乙基纤维素聚合物的软化点的温度,并且开始转子的旋转。转子的旋转可以在使可密封容积密封前或后的任何方便时刻开始。在使可密封容积密封后,继续在高于乙基纤维素聚合物的软化点的温度下旋转转子,直到形成含有乙基纤维素聚合物的分散颗粒(如下所述)的组合物。在其它实施例(“两次”实施例)中,将包括乙基纤维素聚合物、脂肪酸、和任选的额外成分的材料放在混合器中。当将材料放在混合器中时,混合器可以在15℃至99℃的任何温度下。然后将乙基纤维素聚合物、脂肪酸、和任选的额外成分加热到足以制造足够柔软以允许转子旋转的材料混合物的温度,并且使转子旋转。直到此点,可密封容积可能被密封或可能未被密封。然后如果没有密封,则使可密封容积密封。然后按照方便的任何顺序进行以下步骤:将水溶性碱的水溶液注入到密封的可密封容积中(即,进行步骤(c));将可密封容积中的材料加热到高于乙基纤维素聚合物的软化点的温度,并且开始旋转转子。在使可密封容积密封后,继续在高于乙基纤维素聚合物的软化点的温度下旋转转子,直到形成含有乙基纤维素聚合物的分散颗粒(如下所述)的组合物。不管使用单次实施例或两次实施例或一些其它实施例,优选的步骤是进行该方法,以先制造浓缩分散体。浓缩分散体是含有其中固相占基于组合物的总重量的组合物的70重量%或更多的分散颗粒的组合物。优选地是,虽然乙基纤维素聚合物、脂肪酸、水、水溶性碱、和任选的额外成分全存在于混合器中,并且转子正在旋转,同时材料处于高于乙基纤维素聚合物的软化点的温度下,但是已经选择了乙基纤维素聚合物、脂肪酸、水、水溶性碱、和任选的额外成分的量以使水的量是基于混合器中材料的总重量的30重量%或更少。更优选地,水的量是基于混合器中材料的总重量的26重量%或更少;更优选地23重量%或更少。优选地,当制备浓缩分散体时,在进行步骤(e)(如上所述)后,将额外水(标记为“稀释水”)加入组合物中。优选地,在加入稀释水后,混合器中的组合物仍然是本发明的组合物,固体含量是基于组合物的重量的5重量%或更高;更优选地10重量%或更高。优选地,在加入稀释水后,混合器中的组合物仍然是本发明的组合物,并且固体含量是基于组合物的重量的40重量%或更低;更优选地30重量%或更低。优选地,在制备含有乙基纤维素聚合物的分散颗粒的组合物后,将组合物从混合器中除去。本发明的方法是分批方法。也就是说,将各种成分放入混合器中,完成本发明的方法,制备一定量的含水组合物,并在将任何其它成分放入混合器前,将此量的含水组合物从混合器中除去。本发明不包括其中将一些但不是全部的含有乙基纤维素颗粒的分散颗粒的组合物从混合器中除去的方法,然后将另外的乙基纤维素聚合物放入混合器中。本发明的含水组合物优选地具有12或更低;更优选地11或更低;更优选地10或更低的ph。本发明的含水组合物具有8或更高的ph。本发明的含水组合物中的分散粒子优选地具有3微米或更小;更优选地2微米或更小的体积平均粒径。本发明的含水组合物中的分散粒子优选地具有50nm或更大;更优选地100nm或更大的体积平均粒径。使用激光衍射测量粒径。合适的仪器是coultertmls-230或coultertmls-13-320粒度分析仪(贝克曼库尔特(beckmancoultercorporation))。本发明的含水组合物的粘度是在25℃下使用具有在50rpm下旋转的rv2或rv3锭子的布鲁克费尔德(brookfield)rv-ii粘度计测定。选择锭子以给出最接近粘度计扭矩范围中心的扭矩信号。优选地,含水组合物的粘度是100mpa-s或更低;更优选地是80mpa-s或更低;更优选地是60mpa-s或更低;更优选地是40mpa-s或更低;更优选地是30mpa-s或更低。优选地,含水组合物的粘度是1mpa-s或更高。本发明的含水组合物优选地具有基于含水组合物的重量的5重量%或更多;更优选地10重量%或更多;更优选地15重量%或更多;更优选地20重量%或更多的固体含量。本发明的含水组合物优选地具有基于含水组合物的重量的55重量%或更少;更优选地50重量%或更少;更优选地45重量%或更少;更优选地40重量%或更少;更优选地35重量%或更少的固体含量。本发明的含水组合物的优选用途是制备膜。本发明的含水组合物任选地与额外成分混合;将一层本发明的含水组合物施加到表面,并且除去水。所得膜优选地含有基于膜重量的0至5重量%;更优选地0至2重量%;更优选地0至1重量%;更优选地0至0.5重量%的量的残余水。所得膜可用于任何目的。优选的目的是作为药物包衣或食品涂层;更优选地是药物包衣;更优选地是改性释放的药物包衣。制备改性释放的药物包衣的优选方法是提供含有药物的多颗粒制剂,并施加膜的包衣以包覆或包封多颗粒中的每一个。优选的多颗粒由糖或微晶纤维素制成,并将药物作为一层施加到表面或喷涂到表面上。替换地,例如如果多颗粒通过挤出,然后将药物与制成多颗粒的材料的混合物球化而制得,则多颗粒可以含有位于颗粒内部的药物。由本发明的含水组合物制成的膜形成的涂层优选地在50%或更多的颗粒(以数量计)上形成完整的涂层;更优选地,涂层在75%或更多的颗粒上(以数量计)形成完整的涂层。优选地,在90%或更多的颗粒(以数量计)上,涂层覆盖每个颗粒表面面积的75%或更多。合适的多颗粒可以是丸粒、颗粒、粉末、或其它形式。也期望其中将膜用作例如片剂或胶囊的药物剂型上的改性释放的包衣的实施例。当将本发明的含水组合物用于制备膜时,优选地使用增塑剂。当使用增塑剂时,可以在制备组合物的过程期间的任何点将增塑剂加入组合物中。优选地,当使用增塑剂时,将增塑剂与乙基纤维素聚合物同时加入(即,步骤(b)期间)。也想到了制备本发明组合物,然后将增塑剂加入相同的混合器中或在将组合物从制备其的混合器中除出后加入一些其它容器中的实施例。当使用增塑剂时,优选地是选自由以下组成的群组的一种或多种增塑剂:甘油三酸酯、有机酯、具有200或更高的分子量的聚乙二醇、和烷基羧酸;更优选地是柠檬酸三乙酯(tec)、癸二酸二丁酯(dbs)、邻苯二甲酸二乙酯、邻苯二甲酸二丁酯、具有200或更高的分子量的聚乙二醇、和甘油三酸酯;更优选地是柠檬酸三乙酯(tec)、癸二酸二丁酯(dbs)、邻苯二甲酸二乙酯、和邻苯二甲酸二丁酯。当使用增塑剂时,增塑剂的量优选地是基于固相的总干重的10重量%或更多;更优选地15重量%或更多。当使用增塑剂时,增塑剂的量优选地是基于固相的总干重的40重量%或更少;更优选地30重量%或更少。当组合物在多个颗粒上形成涂层时,期望涂层具有良好的膜性能,例如相对高值的杨氏模量、拉伸强度、和最大伸长率。希望这些性质可以通过制备自由膜(即,不粘附到任何基底的膜)并测试自由膜的拉伸性能来测试。希望具有作为自由膜的可接受性能的膜当涂覆到多颗粒上时也将具有可接受的性能。以下是本发明的实例。v平均是体积平均粒径。“d<90%”是90体积%的颗粒低于的直径。粒径“模式”是观察到颗粒群体对直径的曲线中的峰值处的直径。使用coultertmls-230或coultertmls-13-320粒径分析仪(贝克曼库尔特(beckmancoultercorporation))测量粒径。实例1:使用低分子量乙基纤维素聚合物和koh的单次实施例。混合器:如图1所示的混合器。可密封容积的大小是1.9升(4品脱)。以重量计的比例:(74/17/9)74份ethocelstd.10;17份癸二酸二丁酯;9份油酸。碱是koh。先将加热器设定到90℃。使混合器装有196.1gethoceltmstd.10粉末、45.05g癸二酸二丁酯(dbs)、23.85g油酸、20.85g30重量%koh、62mldi(去离子)水。在初始加水期间将碗密封后,以低速率打开刀片以完全润湿粉末。然后利用氮气将碗加压至0.517mpa(75psig),并将浴设定点增加至165℃。该温度设定点最终导致145℃的碗测量温度。在加热期间,混合器缓慢地转动,一旦温度稳定,以最大速率开启混合器,持续30分钟。在此初始混合后,以5ml/min加入稀释水的头200ml,然后剩余483ml以15ml/min加入以得到以负载成分计的26.5%固体的批料。混合器在稀释期间保持最大速率。加入所有水后,停止混合并将加热器设定点降回至90℃。一旦将测量的碗温度降至低于100℃,则将压力释放出来并回收碗中的材料,如下分析。加载到碗中的固体几乎完全转化成浅灰色分散体(回收965g材料,加载1000g)。30.75%固体,ph=8.73,粒径模式=106nm实例2:利用高分子量乙基纤维素聚合物和koh的两次方法。混合器和比例与实例1相同。先将加热器设定到90℃并装有196.1gethoceltmstd.20粉末、45.05g癸二酸二丁酯(dbs)、23.85g油酸。然后将混合器密封,利用氮气加压至0.517mpa(75psig),并加热到165℃的设定点,得到145℃的碗测量温度。此时,加入20.85g23重量%koh(1.2:1的碱:酸当量比)和62ml去离子(di)水,同时混合器缓慢地转动。一旦加入所有的水和碱,将混合器以最大速率(75rpm)打开,持续30分钟。在此初始混合后,以5ml/min加入稀释水的头200ml,然后剩余305ml以15ml/min加入以得到基于负载成分的约38重量%固体的批料。混合器在稀释期间保持最大速率。加入所有水后,停止混合并且将加热器设定点降回至90℃。一旦将测量的碗温度降至低于100℃,则将压力释放出来并且回收碗中的材料,如下分析。在排空混合器时存在少量固体物质。粒径分布在约100nm处具有主峰和少量4-10微米范围内的较大尺寸的物质。分析结果:38.5%固体,ph=8.83,粒径模式=106nm,v平均=0.704微米,d<90%=0.210微米。实例3:使用高分子量乙基纤维素聚合物和氨的两次方法。混合器和比例与实例1相同。碱是氨。先将加热器设定到90℃并装有196.1gethoceltmstd.20粉末、45.05g癸二酸二丁酯(dbs)和23.85g油酸。将混合器密封并加压至0.517mpa(75psig),并且将浴温度设定到165℃,其最终得到145℃的碗测量温度。此时,将混合器打开至低速,将浴设定点降至155℃,将46.21ml28%氨溶液(8:1的碱:酸当量比)和44.9ml水(22重量%水)在利用注射泵缓慢混合下被输送到碗中。加入水和碱后,将混合器设定为其最大速度并运行30分钟。在此初始混合后,以5ml/min加入稀释水的头200ml。在此第一剂量的稀释水后,混合碗中有相当多的泡沫。然后,以15ml/min加入494ml稀释水,得到基于负载成分的26.5%固体目标的批料。混合器在稀释期间保持最大速率。加入所有水后,停止混合并将加热器设定点降回至90℃。一旦将测量的碗温度降至低于100℃,则将压力释放出来并回收碗中的材料。碗中的材料主要是含水分散体,具有一些易碎的固体和一些泡沫。从1034g负载物中回收837g材料(80.9%回收率)。这种分散体的分析如下:ph=9.45,固体=14.72%,v平均=35.39(在200nm处1个峰,2-200微米之间多个峰)实例4:利用高分子量的乙基纤维素聚合物和氨的单次方法混合器和比例与实例1相同。碱是氨。先将加热器设定到90℃并装有196.1gethoceltmstd.20粉末、45.05g癸二酸二丁酯(dbs)、23.85g油酸、46.21g28%氨水溶液、和44.9mldi水。然后将混合器密封,利用氮气加压至0.517mpa(75psig),并缓慢混合数分钟以润湿干燥成分。将碗加热器打开至165℃的设定点,得到145℃的测量的碗温度。一旦达到该温度,则以最大速率(75rpm)打开混合器,持续30分钟。在此初始混合后,以5ml/min加入稀释水的头350ml,然后以15ml/min加入剩余的250ml,得到基于负载成分的27.7重量%固体的批料。混合器在稀释期间保持最大速率。加入所有水后,停止混合并将加热器设定点降回至90℃。一旦将测量的碗温度降至低于100℃,则将压力释放出来并回收碗中的材料,分析如下。在最后几磅/平方英寸的减压期间,出现大量的泡沫,可能来自溶解的过量氨。粒径分布在约350nm处具有主峰和少量2-50微米范围内的较大尺寸的物质。分析结果如下:固体=28.5%,ph=8.71,粒径模式=358nm,v平均=4.18微米,d<90%=10.32微米。实例5:利用低分子量的乙基纤维素聚合物和氨(4:1)的两次方法混合器和比例与实例1相同。碱是氨。先将加热器设定到90℃并装有196.1gethocelstd.10粉末、45.05g癸二酸二丁酯(dbs)和23.85g油酸。将混合器密封并加压至0.517mpa(75psig),并将浴温度设定到165℃,其最终得到145℃的碗测量温度。此时,将混合器打开至低速,将浴设定点降至155℃,利用isco注射泵将23.1ml28%氨溶液(4:1的碱:酸当量比)和59.85ml水输送到碗中。加入水和碱后,将混合器设定为其最大速度并运行40分钟。在此初始混合后,以5ml/min加入稀释水的头200ml,然后,以15ml/min加入剩余的460ml,得到基于负载成分的26.5重量%固体的批料。混合器在稀释期间保持最大速率。加入所有水后,停止混合并将加热器设定点降回至90℃。一旦将测量的碗温度降至低于100℃,则将压力释放出来并回收碗中的材料。回收的材料不是聚合物颗粒在水中的分散体。回收的材料是具有少量游离水的柔软的脆性固体。实例6:利用低分子量的乙基纤维素聚合物和氨(3.5:1)的单次方法。混合器和比例与实例1相同。碱是氨。先将加热器设定到90℃并装有196.1gethoceltmstd.10粉末、45.05g癸二酸二丁酯(dbs)、23.85g油酸、20.22g28重量%氨水(3.5:1的碱:酸当量比)、和62mldi水。在初始加水期间将碗密封后,以低速率打开刀片以完全润湿粉末。然后利用氮气将碗加压至0.517mpa(75psig),并将浴设定点增加至165℃。该温度设定点最终导致145℃的碗测量温度。在加热期间,混合器缓慢地转动,一旦温度稳定,以最大速率开启混合器,持续30分钟。在此初始混合后,以5ml/min加入稀释水的头200ml,然后以15ml/min加入剩余的468ml,得到基于负载成分的26.5重量%固体的批料。混合器在稀释期间保持最大速率。加入所有水后,停止混合并将加热器设定点降回至90℃。一旦将测量的碗温度降至低于100℃,则将压力释放出来并回收碗中的材料。回收的材料不是聚合物在水中的分散体。碗中的材料完全转化为具有少量游离水的柔软的脆性固体。放置过夜后,此游离水被吸收到固体中,冷却时变得更硬。实例7:利用高分子量的乙基纤维素聚合物和氨(2.5:1)的单次方法。混合器和比例与实例1相同。碱是氨。先将加热器设定到90℃并装有196.1gethoceltmstd.20粉末、45.05g癸二酸二丁酯(dbs)、23.85g油酸、14.55g28重量%氨水溶液(2.5:1的碱:酸当量比)、57.8mldi水。在初始加水期间将碗密封后,以低速率打开刀片以完全润湿粉末。然后利用氮气将碗加压至0.517mpa(75psig),并将浴设定点增加至175℃。该温度设定点最终导致155℃的测量的碗温度,在该点以最大速率开启混合器,持续30分钟。在此初始混合后,以10ml/min加入稀释水的头300ml,然后以15ml/min加入剩余375ml,得到基于负载成分的26.5%固体的批料。加入所有水后,停止混合并将加热器设定点降回至90℃。一旦将测量的碗温度降至低于100℃,则将压力释放出来并回收碗中的材料。回收的材料是灰色粉末状固体和大量深棕色水。从此水的固体%测量值(0.4%),我们可以看出,很少的负载固体处于水相中。实例8:parr混合器,低分子量乙基纤维素聚合物,koh比例与实施例1相同。碱是koh。反应容器是装有cowles刀片的300mlparr容器(型号4560)反应器。cowles刀片具有约2"直径。parr容器最初装有47.60gethocelstd.10粉末、11.02g癸二酸二丁酯(dbs)、5.81g油酸、5.10g30重量%koh、16.18mldi水。然后将容器密封,将加热器温度设定为145℃,将cowles混合叶片打开至低速。不向反应器施加外部压力。在加热期间,混合器缓慢转动,一旦温度稳定下来,以最大速率(约1800rpm)打开混合器,持续30分钟。在此初始混合后,利用高效液相色谱(hplc)泵以5ml/min加入稀释水的头47.5ml,然后以15ml/min加入剩余的120ml,得到基于负载成分的26.05重量%固体的批料。混合器在稀释期间保持最大速率。加入所有水后,去除加热套以允许容器冷却同时仍以最大速率混合。一旦测量的容器温度降至50℃以下,则回收容器中的物质,分析如下。装入碗中的固体完全转化为浅灰色分散体。测得的固体%为24.56%,略低于理论固体。比较实例c-ex(在挤出机中制备的分散体)挤出是一种连续方法并且不属于本发明。使用以下组分和条件制备基于挤出机的比较ethocel分散体:组分1:ethocelstd.20组分1进料速率:42.0g/min组分2:癸二酸二丁酯组分2进料速率:9.6g/min组分3:油酸组分3进料速率:5.1g/min初始水进料速率:14.6g/min碱:28重量%氨水溶液碱进料速率:1.9g/min稀释水进料速率:140g/min聚合物熔体区的挤出机温度:145℃挤出机速度:470rpm通用程序如下。使用受控速率进料器;使用如上所述的以克/分钟(g/min)计的进料速率将组分1进料到25毫米(mm)直径的双螺杆挤出机中。将组分2和3进料到挤出机的熔体区中的液体喷射器中并与组分1组合以形成液体熔体材料。使挤出机的温度曲线逐渐升至约145℃。将水和碱混合在一起并以上述速率进料到挤出机,以在初始水引入位置进行中和。然后利用受控速率泵将稀释水以上述速率送入挤出机的稀释区。挤出机速度为约470转/分钟(rpm)。在挤出机出口处,使用背压调节器将挤出机机筒内的压力调节到适于减少蒸汽形成的压力(通常,压力为约2mpa(约300psia))。使用coulterls-230粒径分析仪(可购自贝克曼库尔特(beckmancoultercorporation))测量含水分散体的固体颗粒的粒径。实例9:所选实例的结果ex=实例编号amm=氨ecp=乙基纤维素聚合物低=ethoceltmstd10高=ethoceltmstd20碱amt.=碱:酸当量比cont=连续方法模式=粒径模式,单位为微米v平均=体积平均粒径,单位为微米d<90%=90%的颗粒低于的直径,以体积计无分散体=没有形成分散体rib=图1所示的带状混合器imp=带叶轮的parr反应器extr=挤出机实例混合器碱ecp碱amt.次模式v平均d<90%1ribkoh低1.4:110.106μm0.491μm0.214μm2ribkoh高1.4:120.106μm0.704μm0.210μm3ribamm高8:1298.5μm35.4μm122μm4ribamm高8:110.358μm4.185μm10.32μm5ribamm低4:12无分散体无分散体无分散体6ribamm低3.5:11无分散体无分散体无分散体7ribamm高2.5:11无分散体无分散体无分散体8impkoh高1.4:110.117μm37.44μm140μmc-ex.extramm高1.8:1cont0.141μm0.144μm0.230μm本发明的方法能够使用带式混合器或叶轮式混合机制得分散体;带式混合机通常制得较小粒径的分散体。koh以低得多的碱摩尔量制得分散体。利用低分子量和高分子量的乙基纤维素聚合物制备良好的分散体。在氨样品中,在较高量的碱下制得较好的分散体。比较实例c-a:四脚混合器四脚混合器例如在wo2008/052112中有所描述。四脚混合器是一种装置,其中将布置成正方形的平行或稍微倾斜的四个圆柱形杆与要混合的成分一起插入容器中,并且杆各自围绕其自身的轴线旋转。当杆旋转时,它们不会在杆本身的体积以外的空间中遇到任何点。因此,杆的旋转不会导致杆在先前被混合材料占据的空间中遇到任何点。因此,四脚混合器的%vunc为100%。不对测试的四脚混合器加热,因此使用硅油和表面活性剂代替乙基纤维素和脂肪酸评估四脚混合器的有效性,在环境温度(约23℃)下测试。硅油是具有100,000mpa-s(100,000cps)的粘度的硅油或具有300,000mpa-s(300,000cps)的粘度的硅油。表面活性剂是不同比例的来自shell的neodoltm23-65乙氧基化物与水的混合物。硅油与表面活性剂/水混合物的重量比是94:6。根据两种不同的方案使脚旋转:在一种方案中,所有杆沿相同方向旋转;在另一个方案中,相邻的杆沿相反的方向旋转。将混合物处理5分钟或10分钟。在四脚混合器中制得浓缩分散体;然后将浓缩的分散体稀释,并使用库尔特(coulter)计数器分析粒径。在使用四脚混合器的各种配方和工艺变量组合中,几种得到稳定的分散体,但是由它们中任一种制得的最小粒径具有2微米的体积平均粒径。比较实例c-b:硅油在图1的混合器中。将比较实例c-a中所述的硅油制剂在与实例1相同的混合器中加工。成分如下:油-100=具有100mpa-s的粘度的硅油油-300=具有300mpa-s的粘度的硅油surf-1=来自shell的neodoltm23-65表面活性剂surf-2=来自huntsman的empiricoltmesb70表面活性剂使用两种不同的混合步骤。在“预混合”步骤中,将水和表面活性剂混合在一起,然后将其与硅油一起放入混合器中(将表面活性剂和水放在括号中,表示在下表中)。在“无预混合”步骤中,将硅油、表面活性剂、和水全部混合在混合器中。材料的量(重量份)和所得分散体的粒径如下表所示。粒径是体积平均直径。混合物混合条件大小(μm)90油-1/6surf-1/4水在43rpm下5分钟2.190油-1/6surf-1/4水在120rpm下10分钟0.795油-1/3surf-1/2水在43rpm下5分钟1.495油-1/3surf-1/2水在120rpm下10分钟0.890油-1/10(surf-1/水60:40)在43rpm下5分钟4.090油-1/10(surf-1/水60:40)在120rpm下10分钟1.695油-1/5(surf-1/水60:40)在43rpm下5分钟1.995油-1/5(surf-1/水60:40)在120rpm下10分钟0.894.8油-2/5.8surf-2/3.5水在120rpm下10分钟1.8比较实例c-a和c-b的结果显示了c-b中使用的双带状混合器优于c-a中使用的四脚混合器。在c-b中制得的几乎所有的分散体具有比c-a制得的最小结果(2微米)更小的粒径。希望如果使用相同的两个混合器来制造乙基纤维素聚合物分散体,则双带状混合器将再次制得比四脚混合器更小的粒径。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1