一种治疗方法和用于所述治疗方法中的化合物与流程
发明领域
本发明总体上涉及一种治疗性或预防性治疗局部缺血诱导的心肌组织损伤、特别是局部缺血再灌注诱导的心肌组织损伤的方法。更具体地说,本发明涉及一种通过选择性地上调fpr1介导的erk信号传导来降低哺乳动物中局部缺血诱导的心肌组织损伤的程度的方法。本发明的方法尤其可用于降低与特征在于心肌局部缺血或心肌局部缺血和再灌注的病状(如由动脉粥样硬化性动脉闭塞或血凝块诱导的动脉闭塞引起的急性心肌梗塞)相关的心肌组织损伤的程度和/或严重性。
发明背景
本说明书中被作者提及的出版物的文献著录细节按字母顺序收集在说明的结尾处。
在本说明书中对任何现有出版物(或源自其的信息)或对任何已知内容的引用,不被并且不应被认为是认同、承认或任何形式的暗示所述现有出版物(或源自其的信息)或已知内容形成本说明书所涉及致力领域的公知常识的一部分。
冠状动脉闭塞后急性心肌梗塞(ami)是发达国家的单一最大死亡原因。大约10%的患者由于心律失常或心脏破裂而在头几天内突然死亡,而50%的患者将在2年内发展心力衰竭。
冠状动脉闭塞导致缺氧并导致心肌细胞坏死,其引发可在3个阶段中考虑的复杂响应:起始期、急性炎症期和修复期(frangogiannis,pharmacolres.2008,58:88-111)。最终,此过程导致左心室(lv)重塑(一种维持心输出量的补偿机制)的纤维化。ami对lv功能的有害影响不仅取决于梗塞的面积,而且取决于炎症和修复的平衡。
在局部缺血数分钟内,先天性免疫系统通过补体活化、toll样受体4(tlr4)表达和活性氧种类(ros)产生引发。在动物模型中,补体的抑制减少炎症响应并改善lv功能,但是这些益处尚未在人体试验中重现(armstrong等人2007,jama297:43-51;granger等人circulation.2003,108:1184-1190;mahaffey等人circulation.2003,108:1176-1183)。通过抑制tlr4预防起始期在动物模型中也是有效的,但尚未在人中进行测试(timmers等人2009,jamcollcardiol.53:501-510)。靶向ros不太可能是有用的,因为ros是在局部缺血的几分钟内产生的(lefer和granger,amjmed.2000,109:315-323)。
急性炎症期在局部缺血的数小时内开始,并且其特征在于嗜中性粒细胞和m1样巨噬细胞的细胞浸润,连同大量“促炎性”趋化因子和细胞因子。在前24小时内迅速积聚的嗜中性粒细胞是有助于清除死细胞和碎片的基质金属蛋白酶(mmp)的丰富来源。m1样巨噬细胞是急性炎症期期间存在的其他主要的细胞类型。迁移至局部缺血部位是由ccr2与高水平的mcp-1(ccl2)的结合介导的。粒细胞-巨噬细胞集落刺激因子(gm-csf)促进由m1样细胞产生炎性细胞因子(tnf-α、il-6和il-1β)(fleetwood等人2007,jimmunol.178:5245-5252)。
已经测试了用于抑制炎症期的几种策略,取得了不同程度的成功。靶向特异性炎性细胞因子已经产生了混合结果。tnf-α缺陷型小鼠具有改善的ami后的lv功能,然而,施用抑制性可溶性tnf受体损伤lv功能(monden等人2007,cardiovascres.73:794-805;sun等人2004,circulation.110:3221-3228)。类似地,il-1的抑制已显示有益的或有害的作用(hwang等人2001,jamcollcardiol.38:1546-1553;suzuki等人2001,circulation.104:i308-i303)。这些结果表明靶向个别细胞因子可能不是有用的。
用于心肌梗塞患者的当前治疗是用血栓裂解剂或介入性手术进行血管再生。这些治疗集中于恢复缺血组织的血流,以防止组织坏死并保护器官功能。美国心脏协会(americanheartassociation)针对呈现心肌梗塞的患者的最新建议包括在患者呈现后随后30分钟内的纤维蛋白裂解和患者呈现后90分钟时的直接经皮冠状动脉介入术(pci)(masoudi等人,2008;anderson等人,2011)。引入包括阿司匹林、抗血小板药物、ace抑制剂、arb、β-肾上腺素受体拮抗剂、ca2+-通道拮抗剂、抗血栓裂解剂和糖蛋白(gp)iib/iiia抑制剂的疗法已经改善了心肌梗塞后的结果。尽管这些药物疗法中的大多数仍然在临床使用中,但心肌梗塞和由此产生的心力衰竭仍然是死亡和残疾的主要原因。
然而,在一段时间的局部缺血后恢复血流可能引发进一步的心肌损伤,其被称为再灌注损伤。再灌注损伤的表现除了显著心肌细胞损失外还包括心律失常、心肌顿抑、微血管功能障碍和收缩功能不完全恢复。已经表明,活性氧种类(ros)的过度产生、细胞内ca2+超负荷和炎性细胞浸润是心肌局部缺血-再灌注(i-r)损伤的最重要特征。
在缺血心肌的再灌注期间,心肌坏死通过释放内源性分子引发无菌炎性响应,这已被指定为损伤相关的分子模式(damage-associatedmolecularpatterns)(damps)(chen,g.y.和g.nunez,sterileinflammation:sensingandreactingtodamage.natrevimmunol,2010.10(12):p.826-37)。这些分子由也参与微生物病原体识别和炎性响应的受体感测。已牵涉于心肌局部缺血再灌注损伤中的这样一组受体是toll样受体(tlr)(arumugam,t.v.,等人,toll-likereceptorsinischemia-reperfusioninjury.shock,2009.32(1):第4-16页;chao,w.,toll-likereceptorsignaling:acriticalmodulatorofcellsurvivalandischemicinjuryintheheart.amjphysiolheartcircphysiol,2009.296(1):第h1-12页)。tlr4(革兰氏阴性细菌细胞壁组分脂多糖(lps)的受体)在心肌细胞上表达,并且是内毒素血症中lps诱导的心肌功能障碍的原因(cha,j.,等人,cytokineslinktoll-likereceptor4signalingtocardiacdysfunctionafterglobalmyocardialischemia.annthoracsurg,2008.85(5):第1678-85页;tavener,s.a.和p.kubes,cellularandmolecularmechanismsunderlyinglps-associatedmyocyteimpairment.amjphysiolheartcircphysiol,2006.290(2):第h800-6页)。
尽管认识到心脏局部缺血-再灌注损伤的生理机能,并且即使存在基于心肌缺血性-再灌注损伤的基本发病机制的干预措施,如抗氧化剂和ca2+通道阻断剂,但是临床研究仍显示有限的成功。因此,有效地且可重现地治疗这种病状以便减少心脏损伤的方法仍然是难以捉摸的。因此,对于开发治疗经历心肌局部缺血发作的患者以便最小化由长期局部缺血引发并且当再灌注发生时可使损伤更严重的心肌组织损伤的方法存在持续需要。目的是在心肌细胞活力和收缩功能方面维持和/或恢复心肌功能性。
为此,糖皮质激素(gc)-调控的抗炎介质膜联蛋白-a1(anx-a1)的治疗潜力最近已经在一系列全身性炎性病症中得到认可。anx-a1结合并活化为七跨膜g蛋白偶联受体(gpcr)家族的成员的甲酰肽受体(fpr)家族,以抑制嗜中性粒细胞活化、迁移和浸润。
人fpr家族的fpr2成员(也包括fpr1和fpr3)现在被鉴别为负责anx-a1、其n-末端肽和lxa4(ye等人,2009)和大量其他配体的一些生物学活性的受体。fpr1和fpr2受体广泛分布在组织和不同细胞类型中,在涉及炎症过程的细胞类型上最突出地表达,而fpr3被认为仅在树突细胞中高度表达。然而,fpr2和fpr3共享之70%水平的序列同源物(ye等人,2009)。关于anx-a1及其肽模拟物(ac2-26,cgen-855a)的心脏保护作用的研究主要集中于其抗炎作用作为在i-r损伤后保持心肌活力的机制。然而,现在还有证据表明anx-a1对心肌的直接保护作用,其与体外炎性细胞无关。因此,除减少嗜中性粒细胞浸润(抗炎)外,anx-a1保持心肌细胞活力和收缩功能的能力因此突出了anx-a1作为改善心肌梗塞后结果的临床方法的潜在治疗效用。然而,这种方法仍在进行广泛的研究,并且其方法尚未在诊所展开。因此,对于开发用于心肌i-r损伤的新治疗方案的需要持续。
已经观察到,虽然anx-a1-fpr2相互作用例示了一种可能的治疗心肌梗塞患者的手段,但仍然存在与这种相互作用相关的不期望的副作用。具体地说,fpr介导的信号传导可导致再灌注组织中的局部化ca2+超负荷,且由此诱导细胞死亡。然而,在得到本发明的工作中,已经鉴别了在诱导fpr介导的信号传导方面出人意料地表现出高度选择性功能性的fpr激动剂。具体地说,除了选择性地诱导fpr1信号传导(与fpr2信号传导相反)外,本发明的化合物还实现功能偏向性,在于仅fpr1-erk/akt介导的信号传导途径被上调,出人意料地这选择性地活化心肌细胞存活机制而无通常在fpr活化情况下观察到的细胞内ca2+动员的伴随活化。此外,fpr1介导的信号传导已被确定为不仅实现心肌细胞收缩功能的恢复,而且保持心肌细胞活力,从而不仅提供anx-a1/fpr2相关心肌细胞存活作用的有效替代方案,而且事实上显著改善效应机制,与fpr2活化不同,所述效应机制允许仅选择性上调erk/akt信号传导,而不选择性上调细胞内ca2+动员。因此,本发明经由fpr1-erk/akt信号传导的选择性上调,相对于关于治疗心肌梗塞、且具体地说局部缺血-再灌注损伤所先前已知的提供出人意料的和优异的结果。因此,本发明使得能够开发一种治疗方法,关于所述治疗方法,ca2+超负荷的不期望的副作用和潜在地还有ca2+-触发的促炎性作用已被最小化,同时仍然使用单一化合物上调心肌收缩功能和活力,所述化合物不仅选择性地上调fpr1活性,而且使诱导的受体信号偏斜为细胞保护性erk/akt介导的响应。
发明概述
在本说明书和随附的权利要求书的全文中,除非上下文另有要求,否则词语“包含/包括(comprise)”和如“包含/包括(comprises)”和“包含/包括(comprising)”的变化形式应理解为暗示包括所陈述的整体或步骤或者整体或步骤的组,但不排除任何其他整体或步骤或者整体或步骤的组。
如本文所用,术语“源自”应被认为指示特定整体或整体的组来源于所指定的物种,但不一定直接从所指定的来源获得。此外,除非上下文另外明确规定,否则如本文所用,单数形式“一个/种(a/an)”和“所述”包括复数个指示物。
除非另外定义,否则本文中所用的所有技术和科学术语均具有如本发明所属领域的普通技术人员通常所理解的相同含义。
符号“-”是指单键,并且“=”是指双键。当描绘或描述化学结构时,除非另外明确陈述,否则假定所有碳均具有氢取代以符合四价。本领域的普通技术人员应理解,以上提及的描述性技术在化学领域中是常见的以对另外复杂结构的描述提供简洁和简单。
如果将基团“r”描绘成在环系统上“浮动”,例如在下式中:
那么除非另外定义,否则取代基“r”可存在于环系统的任何原子上,假定置换来自一个环原子的所描绘、所暗示或明确定义的氢,只要形成稳定结构即可。
本发明的一个方面涉及一种使哺乳动物中局部缺血诱导的心肌组织损伤的程度最小化的方法,所述方法包括向所述哺乳动物施用有效量的结构(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(l)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物。
另一方面,提供一种使哺乳动物中局部缺血诱导的心肌组织损伤的程度最小化的方法,所述方法包括向所述哺乳动物施用有效量的结构(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(l)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物,并且其中所述组织损伤是细胞活力和收缩功能丧失。
本发明的另一方面提供一种使哺乳动物中心肌梗塞相关组织损伤的程度最小化的方法,所述方法包括向所述哺乳动物施用有效量的结构式(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(l)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物。
本发明的另一方面涉及一种使哺乳动物中局部缺血-再灌注诱导的心肌组织损伤的程度最小化的方法,所述方法包括向所述哺乳动物施用有效量的结构(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(l)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物。
仍然另一方面,提供一种治疗性或预防性治疗哺乳动物中以局部缺血诱导的心肌组织损伤或局部缺血-再灌注诱导的心肌组织损伤为特征的病状的方法,所述方法包括向所述哺乳动物施用有效量的结构(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(1)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物。
根据前述方面和实施方案,结构(i)的化合物可以是
其中
(a)x是ch2;
y是ch2conh;以及
r1是ch3;
或
(b)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(c)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(d)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(e)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(g)x是ch2;
y是ch(ch3)conh
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;
或
(i)x是ch2;
y是ch(ch3)conh
r1是ch3;
其中r2是氢且r3是对-溴,并且r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基;
或
(j)x是ch2;
y是ch(ch3)conh
r1是ch3;
其中r2和r3独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基,并且r4是氢,且r5是间-甲氧基;
或
(k)x是ch2;
y是ch(ch3)conh
r1是ch3;
其中r2是氢且r3是对-溴,并且r4是氢且r5是间-甲氧基;
或其药学上可接受的盐、溶剂化物或立体异构体。
结构(i)的化合物包括但不限于:
以及其药学上可接受的盐、溶剂化物或立体异构体。
本发明的另一方面涉及如上文所定义的化合物在制造用于使哺乳动物中局部缺血诱导的心肌组织损伤的程度最小化的药物中的用途。
在另一方面,提供如上文所定义的化合物在制造用于使哺乳动物中局部缺血-再灌注诱导的心肌组织损伤的程度最小化的药物中的用途。
本发明的另一方面涉及一种鉴别调节靶受体的活性的候选化合物的方法,所述方法包括:
a)使受体与候选化合物接触,
b)确定所述候选化合物与所述受体的结合;
c)选择活化所述受体的候选化合物;
其中所述靶受体是甲酰肽受体亚型fpr1;以及所述候选化合物使所述fpr1亚型活化至比内源性配体或底物对所述fpr1亚型的活化更高的程度。
优选地,所述化合物选择性地活化erk/akt信号传导。
附图简述
图1是膜联蛋白-a1模拟物对经受缺氧-复氧的新生儿心肌细胞上的ldh(a,b)和ctni(c,d)释放的作用的图形表示。使心肌细胞经受6小时缺氧,然后48小时复氧。处理膜联蛋白-a1n-末端肽ac2-26(1μm)和小分子模拟物(bidi-001和bidi-002,1μm)对于缺氧和复氧的全部持续时间(a,c)或仅在再灌注期间(b,d)存在。
图2是稳定表达人fpr1(a)或fpr2(b)受体的flpln-cho细胞中激动剂刺激(fmlp,bidi-001和bidi-002)对fpr介导的[ca2+]i动员的作用的图形表示。数据点表示来自一式三份进行的3-7次实验的由atp引发的[ca2+]i动员的平均值±sem的百分比。
图3是稳定表达人fpr1(a)或fpr2(b)受体的flpln-cho细胞中激动剂刺激(fmlp,bidi-001和bidi-002)对fpr介导的perk1/2的作用的图形表示。数据点表示从一式三份进行的4-6次实验收集的由10%fbs±sem引发的erk1/2磷酸化的平均值±sem的百分比。
图4是稳定表达人fpr1(a,b)或fpr2(c,d)受体的flpln-cho细胞中激动剂刺激(fmlp,bidi-001和bidi-002)对fpr介导的pakt1/2/3(ser473)和pakt1(thr308)的作用的图形表示。数据点表示从一式三份进行的4-6次实验收集的由10%fbs±sem引发的akt磷酸化的平均值±sem的百分比。
图5是稳定表达人fpr1(a)或fpr2(b)受体的flpln-cho细胞中激动剂刺激(fmlp,bidi-001和bidi-002)对fpr介导的对毛喉素(forkskolin)刺激的camp积聚的抑制的作用的图形表示。数据点表示从一式三份进行的3-4次实验收集的毛喉素响应的平均值±sem的百分比。
图6是稳定表达人fpr1(a)或fpr2(b)受体的flpln-cho细胞中激动剂刺激(fmlp,bidi-001和bidi-002)对fpr介导的[ca2+]i动员、perk1/2、pakt1/2/3、以及camp积聚的抑制的偏向性信号传导的作用的示意性图示。
图7是描绘肽和小分子anx-a1模拟物对24小时i-r损伤后心脏坏死的作用的图像。a)使来自代表性小鼠的lv横切片经受40分钟局部缺血,然后在不同处理组中进行24小时再灌注:媒介物、ac2-26或小分子模拟物(bidi-001,bidi-002)。在用伊文思蓝且然后ttc染色后,三个不同的区可见。染色为深蓝色、白色和红色的区域分别代表非风险区、梗塞区和缺血性但非梗塞区。危险区包括红色和白色区域。b)在不同处理组中经受心肌i-r的小鼠的风险区域的合并数据。c)不同处理组中的梗塞范围的合并数据。d)经受i-r的不同处理组中的心肌肌钙蛋白(ctni)的血浆水平的合并数据。数字表示n/组。相对于假手术,#p<0.05;相对于媒介物处理的小鼠,*p<0.05。单因素方差分析与图基事后检验(tukeypost-hoctest)。数据呈现为平均值±sem。
图8是描绘肽和小分子anx-a1模拟物对i-r后48小时i-r诱导的左心室形态的变化的作用的图像。a)在i-r后48小时,来自假手术小鼠、媒介物处理的小鼠、ac2-26处理的小鼠或媒介物和小分子模拟物(bidi-001,bidi-002)处理的小鼠的lv的代表性h&e染色切片。b)关于h&e染色的组织学损伤评分的合并数据。相对于假手术,#p<0.05;相对于媒介物处理的小鼠,*p<0.05。单因素方差分析与图基图基事后检验。数据呈现为平均值±sem。
图9是描绘肽模拟物和小分子anx-a1模拟物对i-r损伤后48小时lv中i-r诱导的嗜中性粒细胞浸润的作用的图像。a)来自假手术、肽ac2-26和小分子非肽模拟物(bidi-001和bidi-002)的lv的代表性染色切片。对于20x(从在20x放大倍数下拍摄的9x9个单一图像拼接),比例尺为500μm,并且对于40x,比例尺为100μm。b)ly-6b.2信号检测的合并数据。相对于假手术,#p<0.05;相对于媒介物处理的小鼠,*p<0.05。单因素方差分析与图基事后检验。数据呈现为平均值±sem。
图10是描绘肽和小分子anx-a1模拟物对i-r损伤后48小时lv中i-r诱导的巨噬细胞浸润的作用的图像。a)来自假手术、肽ac2-26和小分子非肽模拟物(bidi-001和bidi-002)的lv的代表性染色切片。对于20x(从在20x放大倍数下拍摄的9x9个单一图像拼接),比例尺为500μm,并且对于40x,比例尺为100μm。b)cb68+信号检测的合并数据。相对于假手术,#p<0.05;相对于媒介物处理的小鼠,*p<0.05。单因素方差分析与图基事后检验。数据呈现为平均值±sem。
图11是肽和小分子anx-a1模拟物对i-r损伤后48小时i-r诱导的循环白细胞的变化的作用的图形表示。从假手术、肽ac2-26和小分子非肽模拟物(bidi-001和bidi-002)收集的血液中的a)总wbs、b)嗜中性粒细胞、c)淋巴细胞和d)单核细胞的合并数据。相对于假手术,#p<0.05;相对于媒介物处理的小鼠,*p<0.05。单因素方差分析与图基事后检验。数据呈现为平均值±sem。
图12是描绘肽和小分子anx-a1模拟物对i-r损伤后7天之后i-r诱导的心肌纤维化的变化的作用的图像。a)在假手术、肽模拟物ac2-26和小分子非肽模拟物(bidi-001和bidi-002)中用苦味酸天狼猩红(picrosiriusred)染色的梗塞lv的代表性染色切片。b)不同处理组中的心脏纤维化的合并数据。相对于假手术,#p<0.05;相对于媒介物处理的小鼠,*p<0.05。单因素方差分析与图基事后检验。数据呈现为平均值±sem。
图13是fpr激动剂cmpd17b在体内在i-r损伤后24小时减轻心脏坏死的图像和图形表示。(a)在分配至媒介物、cmpd17b或cmpd43(两者均为50mg/kgi.p.)处理组的小鼠中24小时再灌注后的代表性ttc染色的lv横切片。染色为深蓝色、白色和红色的区域分别代表非风险区、梗塞区和缺血性但非梗塞区。(b)aar(计算为总梗塞区加缺血性但非梗塞区,总lv%)、(c)心肌梗塞范围(aar%)和(d)血浆ctni水平的合并数据。结果表示为平均值±sem,其中指示n/组。使用单因素方差分析与图基事后检验,相对于假手术,#p<0.05,###p<0.001且####p<0.0001;相对于媒介物处理的i-r,*p<0.05且***p<0.001。
图14是fpr激动剂cmpd17b在i-r损伤后48小时后减轻心脏炎症的图像和图形表示。(a)i-r后48小时来自假手术、媒介物和fpr激动剂处理的(cmpd17b或cmpd43,50mg/kg,i.p.)小鼠的lv嗜中性粒细胞含量(使用抗ly-6b.2抗体)的代表性免疫荧光图像。)b)lvly-6b.2阳性免疫荧光的合并数据。(c)i-r后48小时来自假手术、媒介物和fpr激动剂处理的(cmpd17b或cmpd43,50mg/kg,i.p.)小鼠的lv巨细胞含量(使用抗cd68抗体)的代表性免疫荧光图像。(d)lvcd68阳性免疫荧光的合并数据。比例尺:100μm(40x放大倍数)。结果表示为平均值±sem,其中指示n/组。使用单因素方差分析与图基事后检验,相对于假手术,#p<0.05;并且相对于媒介物处理的小鼠,*p<0.05。
图15是fpr激动剂cmpd17b在i-r损伤后48小时减轻全身性炎症的图形表示。在48小时再灌注后假手术、媒介物和fpr激动剂处理的小鼠中a)总wbc、b)淋巴细胞、c)嗜中性粒细胞、d)单核细胞和e)il-1β的循环水平。结果表示为平均值±sem。使用单因素方差分析与图基事后检验,相对于假手术,#p<0.05;相对于媒介物处理的小鼠,*p<0.05。
图16是fpr激动剂cmpd17b在i-r损伤后7天后降低心脏重塑的图像和图形表示。a)在i-r后7天,来自假手术、媒介物和fpr激动剂(cmpd17b或cmpd43,50mg/kgi.p.)处理的小鼠的代表性苦味酸天狼猩红染色的lv切片。b)心脏纤维化的定量(胶原呈红色);放大倍数x200。c)使用cardiotac原位细胞凋亡检测的代表性lv切片(阳性染色的凋亡细胞呈深蓝色,由箭头指示);放大倍数x200。d)凋亡的定量:非凋亡细胞(表示为假手术倍数)。i-r后7天归一化至bw的e)心脏和f)lv重量。结果表示为平均值±sem。使用单因素方差分析与图基事后检验,相对于假手术,#p<0.05,##p<0.01;相对于媒介物处理的小鼠,*p<0.05,***p<0.001,****p<0.0001。
图17是fpr激动剂cmpd17b针对体外心肌细胞损伤响应的保护作用的图形表示。a)小分子fpr激动剂(两者均1μm,在复氧时添加)对h-r后心肌细胞ctni释放的作用。cmpd17b和cmpd43(两者均为10μm)对心肌成纤维细胞、b)促纤维化、c)tgf和c促炎性il-1β表达的tgf-β刺激的作用。结果表示为平均值±sem。使用单因素方差分析与图基事后检验,相对于假手术##p<0.05,且相对于媒介物处理的h-r(心肌细胞)或tgf-β刺激(心肌成纤维细胞),*p<0.05。
图18是稳定表达hfpr1的cho细胞中fpr激动剂的信号传导指纹的图形表示:cmpd17b是hfpr1的偏向性激动剂。fmlp(黑色)、cmpd17b(蓝色)和cmpd43(红色)对gpcr下游的细胞内信号传导中间体的影响。a)erk1/2磷酸化;b)akt1/2/3thr308磷酸化;c)akt1/2/3ser473磷酸化;d)ca2+i;和e)毛喉素刺激的camp积聚的抑制。结果表示为3-4次实验(各自一式三份进行)的平均值±sem。f)“偏向性网”绘制了归一化至参考激动剂(cmpd43)和参考途径(pakt1/2/3thr308)的偏向性因子(δδτ/ka),其来源于图a-e中的结果,表示为平均值。对于ca2+i,*p<0.05cmpd17b对比cmpd43,单因素方差分析,然后图基事后检验。
图19是稳定表达hfpr2的cho细胞中fpr激动剂的信号传导指纹的图形表示。fmlp(黑色)、cmpd17b(蓝色)和cmpd43(红色)对gpcr下游的细胞内信号传导中间体的影响。a)erk1/2磷酸化;b)akt1/2/3thr308磷酸化;c)akt1/2/3ser473磷酸化;d)ca2+i;和e毛喉素刺激的camp积聚的抑制。结果表示为3-4次实验(各自一式三份进行)的平均值±sem。f)示出归一化至cmpd43和参考途径pakt1/2/3thr308的偏向性因子(δδτ/ka)的“偏向性网”,其来源于图a-e中的结果,表示为平均值。对于ca2+i,*p<0.05cmpd17b对比cmpd43,单因素方差分析,然后图基事后检验。
图20是fpr激动剂的偏向性特性的图像,以及它们针对心肌i-r损伤的作用的影响。cmpd17b和cmpd43活化fpr1和fpr2,但是cmpd17b可能通过偏向活化合乎需要的心脏保护性risk信号传导途径(erk1/2,akt1/2/3thr308,akt1/2/3ser473)、同时避开可促成不良后果(例如ca2+i)的途径而提供优异的心脏保护作用。
图21是(a)被鉴别为化合物17b(cmpd17b或bidi-001)的小分子fpr激动剂n-(4-溴苯基)-2-[5-(3-甲氧基苄基)-3-甲基-6-氧代-6h-哒嗪-1-基]-丙酰胺和被鉴别为化合物43(cmpd43或bidi-002)的amgen吡唑啉酮衍生物的化学结构的图像和图形表示。b)为了定量信号传导偏向性,使用激动作用操作模型通过非线性回归对激动剂浓度-响应曲线进行分析以确定对于每种途径,每种激动剂的τ/ka“转导比”,其中em是系统(而不是激动剂)的最大可能响应,ka表示受体的激动剂的功能平衡解离常数,并且τ受体及其后续细胞刺激-响应转导机制的偶联效率。
图22是fpr激动剂cmpd17b在体内在i-r损伤后减轻心脏炎症和纤维化的图像。在i-r后48小时,来自假手术、媒介物和fpr激动剂处理(cmpd17b或cmpd43,50mg/kg,i.p.)的小鼠的a)lv嗜中性粒细胞含量的代表性免疫荧光图像(使用抗ly-6b.2抗体)和b)lv巨噬细胞含量的代表性免疫荧光图像(使用抗cd68抗体)。比例尺:500μm(在20x放大倍数下拼接的9x9个单一图像)。c)在i-r后7天,来自假手术、媒介物和fpr激动剂(cmpd17b或cmpd43,50mg/kgi.p.)处理的小鼠的代表性苦味酸天狼猩红染色的lv切片。比例尺:500μm(放大倍数x12.5)。
图23是体外fpr的心肌细胞表达的图形表示。a)通过实时pcr测定未处理的nrcm中的大鼠fpr(rfpr1、rfpr2和rfpr3)的相对表达(表示为从所使用的最大循环数40减去的阈值循环数ct)。与cmpd17b和cmpd43(两者均为1μm)48小时孵育对b)rfpr1和c)rfpr2的nrcm表达的影响,其表示为相对于媒介物处理的细胞的倍数变化。d通过实时pcr测定的amcf中小鼠fpr(mfpr1和mfpr2)的相对表达。与cmpd17b和cmpd43(两者均为10μm)24小时孵育对emfpr1和fmfpr2的amcf表达的影响,其表示为相对于媒介物处理的细胞的倍数。相对于媒介物,#p<0.05,##p<0.01,###p<0.001且####p<0.0001;相对于tgf-β,*p<0.05,单因素方差分析与图基事后检验。数据表示为平均值±sem。
图24是hfpr亚型中erk1/2磷酸化的图形表示和gpcr激动作用的定量框架的描述。在hfpr转染的cho细胞中,响应于fmlp、cmpd17b、cmpd43和lxa4(各自10μm,n=1)的erk1/2磷酸化的初步时程研究:a)hfpr1;b)hfpr2;c)fpr3。d)hfpr3-cho细胞中每种fpr激动剂的浓度-响应曲线(结果表示为3-4次实验(各自一式三份进行)的平均值±sem)。e)在hfpr亚型中lxa4(示出fmlp用于比较)对erk1/2磷酸化的浓度-响应曲线。
图25是在体外cho细胞中在fpr1下远离ca2+i的bidi-001偏向信号传导的图像和图形表示。
发明详述
本发明部分地基于能够选择性地活化在心肌细胞上表达的fpr1并且此外选择性地活化fpr1-erk/akt介导的信号传导(其用于维持心肌细胞活力并上调收缩功能)的化合物的出人意料的鉴别来预测。然而,这一发现是特别重要的,因为它提供上调心肌细胞活力和收缩功能而无诱导细胞内ca2+动员的伴随不良结果的手段。因此,尽管事实上现有技术方法主要集中于膜联蛋白介导的fpr2活化的心脏保护能力,但现在已经确定fpr1的选择性活化也可实现维持心肌细胞活力和恢复收缩功能两者,但无细胞内ca2+动员的副作用,ca2+动员在心脏局部缺血-再灌注损伤的背景下是非常不希望的并且可导致组织死亡。本发明人首次鉴别了能够实现这种高选择性功能结果的分子。
因此,本发明的一个方面涉及一种使哺乳动物中局部缺血诱导的心肌组织损伤的程度最小化的方法,所述方法包括向所述哺乳动物施用有效量的结构(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(1)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物。
提及“心肌组织”应该被理解为对心脏的组织的提及。这种组织由异质细胞群体组成,并且提及“心肌组织”因此包括对所有这些细胞的提及。这包括例如心肌(即心脏的横纹、不随意肌)以及心脏中存在的其他细胞类型,如血管、神经、结缔组织等。
提及心肌组织“损伤”应被理解为对形成心脏的一部分的一个或多个细胞亚群发生的任何形式的结构、功能或代谢损伤的提及。在不将本发明限于任何一种理论或作用模式的情况下,心肌局部缺血的特征在于对心肌组织供血的限制。由于氧(缺氧)和其他代谢物的输送的合力减少,发生肌细胞坏死,这也被称为心肌梗塞。此事件然后起始复杂响应,有时称为局部缺血级联,其在如下的3个基本阶段发生:
(i)起始期;
(ii)急性炎症期;以及
(iii)修复期。
更具体地说,在局部缺血数分钟内,先天性免疫系统通过补体活化、tlr4表达和活性氧种类(ros)产生引发。急性炎症期在局部缺血的数小时内开始,并且其特征在于嗜中性粒细胞和m1样巨噬细胞的细胞浸润,连同大量“促炎性”趋化因子和细胞因子。在前24小时内迅速积聚的嗜中性粒细胞是有助于清除死细胞和碎片的基质金属蛋白酶(mmp)的丰富来源。m1样巨噬细胞是急性炎症期期间存在的主要细胞类型。迁移至局部缺血部位是由ccr2与高水平的mcp-1(ccl2)的结合介导的。粒细胞-巨噬细胞集落刺激因子(gm-csf)促进由m1样细胞产生炎性细胞因子(tnf-α、il-6和il-1β)(fleetwood2007,同上)。在2-3天后,存在与从促炎性至抗炎性的细胞因子分布的变化相关的从m1样巨噬细胞至m2样巨噬细胞的逐渐转变,以使得新血管形成(血管生成)和纤维化发生。巨噬细胞集落刺激因子(m-csf)促进单核细胞分化为m2样细胞,从而导致抗炎细胞因子如il-10的分泌(fleetwood2007,同上)。然而,心脏纤维化和重塑虽然作为维持心输出量的补偿机制发生,但是对左心室功能是最终有害的。因此,本发明的组织损伤包括但不限于心肌炎症、收缩功能丧失、细胞坏死、细胞凋亡、纤维化、胶原、瘢痕形成、组织重塑、梗塞形成(此后一短语通常用于描述受损伤的心肌组织的质量)和左心室功能。
在一个实施方案中,所述心肌组织损伤是细胞活力和收缩功能的丧失。
根据这一实施方案,提供一种使哺乳动物中局部缺血诱导的心肌组织损伤的程度最小化的方法,所述方法包括向所述哺乳动物施用有效量的结构(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(1)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物,并且其中所述组织损伤是细胞活力和收缩功能丧失。
对使所述组织损伤的“程度最小化”的提及应被理解为对相对于将在未治疗的哺乳动物中发生的组织损伤的严重性或程度,抑制、延缓或以其他方式减轻所述组织损伤的任何一个或多个方面的严重性或程度的提及。因此,所述“最小化”可以是部分的(因为某些程度的损伤确实发生,但并不像否则将会发生的那样广泛)或完全的(因为完全防止损伤),并且可指本文考虑的组织损伤形式中的所有或仅一些。
在一个实施方案中,所述局部缺血诱导的心肌组织损伤是心肌梗塞诱导的组织损伤,并且更具体地说,急性心肌梗塞诱导的组织损伤。
因此,本发明提供一种使哺乳动物中心肌梗塞相关组织损伤的程度最小化的方法,所述方法包括向所述哺乳动物施用有效量的结构式(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(1)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物。
优选地,所述心肌梗塞是急性心肌梗塞。
在不将本发明限于任何一种理论或作用模式的情况下,甲酰肽受体(fpr)属于参与趋化性的一类g蛋白偶联受体。这些受体最初通过它们结合n-甲酰基肽(如通过细菌或宿主细胞降解产生的n-甲酰甲硫氨酸)的能力来鉴别。甲酰肽受体(fpr)属于具有七个疏水跨膜结构域的一类受体。fpr的构象通过几种相互作用来稳定。这些包括arg84-arg205、lys85-arg205和lys85-asp284之间的潜在盐桥形成,其有助于确定跨膜结构域的三维结构;以及与带负电荷的磷酸盐相互作用的带正电荷的残基(arg,lys)。此外,残基arg163可与fpr的第二细胞外环的配体结合口袋相互作用。人甲酰肽受体fpr家族的fpr2成员(也包括fpr1和fpr3)现在被鉴别为负责anx-a1、其n-末端肽和lxa4(ye等人,2009)和大量其他配体的一些生物学活性的受体。fpr1和fpr2受体广泛分布在组织中和不同细胞类型上,在涉及炎症过程的细胞类型上最突出地表达,而fpr3被认为仅在树突细胞上高度表达。然而,fpr2和fpr3共享≥70%的序列同源性水平(ye等人,2009)。关于不同类型的肿瘤细胞的fpr2表达的报道的数量在增加。提及“fpr1”应被理解为对fpr1的所有形式和可从fpr1mrna的选择性剪接产生的任何同种型或其突变体或多态形式的提及。
如上所述,为本发明的主题的心肌组织损伤由局部缺血诱导。“局部缺血”是指心肌缺血性事件,更具体地说是心肌组织的氧合血流量减少。这种减少通常是由于供血限制的发生所致,所述供血限制通常是由于心脏周围的一个或多个动脉的部分或完全阻塞所致。血流量的限制可以是对于全部或仅仅一些心肌组织。它也可能是瞬时限制,其天然地矫正其自身;或者它可能是非瞬时的,因为它需要医疗干预来部分或完全清除。本发明的局部缺血是导致在心肌组织区域中发生一定程度的心肌组织损伤(心肌梗塞)的类型(就其持续时间和闭塞程度而言)。在不将本发明限制于任何一种理论或作用模式的情况下,心肌组织的血流量的限制可起因于广泛范围的事件中的任一种,包括但不限于动脉粥样硬化斑块形成,其使冠状动脉逐渐缩窄(冠状动脉动脉粥样硬化);心外膜冠状动脉中的动脉粥样硬化斑块的破坏,其导致凝血级联,有时导致动脉完全闭塞;动脉粥样硬化斑块的破裂,其也促进血栓(印迹凝块形成);血凝块阻塞,其已从其原始部位移动并且已经行进穿过动脉并停留在服务于心脏的动脉中;心脏内的小动脉和其他小血管的扩散性缩窄;动脉的手术夹紧或血管成形术。
仍然在不将本发明限于任何一种理论或作用模式的情况下,在心肌局部缺血的许多情况下,一定水平的供血将恢复至心肌组织。这可能由于天然发生的事件而发生的,或者其可作为医疗干预的结果发生,所述医疗干预如闭塞性血栓的化学裂解、血管扩张药物的施用、血管成形术、导管插入、插入动脉支架或动脉旁路移植手术。然而,在一段时间的局部缺血后血流的恢复虽然最终是必要的,但实际上可能造成比局部缺血本身更多的损伤。氧重新引入心肌组织尤其导致氧自由基的产生。据认为,在局部缺血期间,血液中氧和营养物质的不存在产生其中循环的恢复通过诱导氧化应激导致更进一步的炎症和氧化损伤的病状。这被称为“再灌注损伤”,并且是对在初始缺氧发生时发生的损伤以及如上所述的随后局部缺血级联的添加。可观察到在再灌注部位发生的损伤的类型包括但不是限于,伴随细胞肿胀和分解的水肿、肌膜破坏、线粒体断裂、收缩带坏死、酶清除、钙超负荷、出血或室性心律失常。
已经确定本发明的方法有效地使局部缺血诱导的心肌组织损伤和潜在更严重的局部缺血-再灌注诱导的心肌组织损伤两者的程度最小化。如上所述,本发明的特别出人意料的和非常显著的方面之一涉及以下事实:本发明的分子不仅选择性地活化心肌细胞fpr1功能性,而且它们还选择性地活化erk/akt介导的信号传导,所述信号传导选择性地促进心脏保护而不是细胞内ca2+动员,从而避免由ca2+超负荷引起的心脏组织死亡的极其不利的副作用。
因此,在一个相关方面,本发明涉及一种使哺乳动物中局部缺血-再灌注诱导的心肌组织损伤的程度最小化的方法,所述方法包括向所述哺乳动物施用有效量的结构(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(l)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物。
在这一方面的一个实施方案中,所述心肌组织损伤是细胞活力和收缩功能的丧失。
在另一个实施方案中,所述局部缺血-再灌注诱导的组织损伤是心肌梗塞诱导的组织损伤,并且更具体地说,急性心肌梗塞诱导的组织损伤。
就结构(i)的化合物而言,并且不将本发明限于任何一种理论或作用模式,如在整个说明书中所用,在本文定义的特定取代基或术语内,可存在两个或更多个相同类型的基团(例如,两个烷基或两个芳基)。除非明确相反地陈述,否则这些基团中的每个可与相同类型的另外每个相同或不同。
术语“取代的”应被理解为是指所指定的基团或部分带有一个或多个取代基。在任何基团可携带多个取代基并且提供多种可能的取代基的情况下,取代基是独立选择的并且不必是相同的。术语“未取代的”是指所指定的基团不携带取代基。术语“任选取代的”是指所指定的基团是未取代的或被一个或多个取代基取代。
任何不对称碳原子可以(r)-、(s)-或(r,s)-构型存在,优选以(r)-或(s)-构型存在,无论哪种是最具活性的。双键或环处的取代基可以顺式-(z)或反式-(e)形式存在。
当前公开的化合物可在分子上含有不对称中心,这取决于各种取代基的性质。在某些情况下,不对称性还可由于与指定化合物的两个芳环相邻的中心键的旋转受限而存在。意图本发明的范围内包括作为分离的、纯的或部分纯化的异构体或其外消旋混合物形式存在的所有异构体(包括对映异构体和非对映异构体),无论是因不对称中心性质还是因如上所述的受限旋转产生。
术语“卤素”或“卤代”是指氟(f,氟-)、溴(br,溴-)、氯(cl,氯-)和碘(i,碘-)原子。
术语“二卤素”、“三卤素”和“全卤素”是指分别各自独立地选自由以下组成的组的两个、三个和四个取代基:氟、溴、氯和碘原子。
术语“烷基”是指直链饱和单价烃基或支链(具有单个或多个支链)饱和单价烃基,例如甲基、乙基、丙基、2-丙基、丁基(包括所有异构形式)或戊基(包括所有异构形式)等。
术语“c1-c6烷基”是指具有一至六个碳原子的直链饱和单价烃基或具有三至六个碳原子的支链(具有单个或多个支链)饱和单价烃基,例如甲基、乙基、丙基、2-丙基、丁基(包括所有异构形式)或戊基(包括所有异构形式)等。
术语“烷氧基”是指-or基团,其中r是如本文所定义的烷基,例如甲氧基、乙氧基、丙氧基、异丙氧基等。
术语“卤代烷基”是指被一个或多个卤素、特别是一至五个卤素原子取代的烷基,例如三氟甲基、2-氯乙基、2,2-二氟乙基等。
术语“卤代烷氧基”是指-or′基团,其中r′是如本文所定义的卤代烷基,例如三氟甲氧基、2,2,2-三氟乙氧基等。
词语“化合物”应被理解为涵盖任何和所有异构体(例如,对映异构体、立体异构体、非对映异构体、旋转异构体和互变异构体)、外消旋体或异构体的任何混合物、前药、以及所述化合物的任何药学上可接受的盐。在化合物、盐等使用复数形式的情况下,这也表示单一化合物、盐等。
本文公开的化合物的“药学上可接受的盐”是指药学上可接受并且具有母体化合物的所需药理活性的盐。应了解药学上可接受的盐是无毒的。关于药学上可接受的盐的另外信息可见于remington’spharmaceuticalsciences,第17版,mackpublishingcompany,easton,pa,1985;或s.m.berge等人,“pharmaceuticalsalts”,j.pharm.sci.,1977,66,1-19中,所述文献均以引用的方式并入本文。
药学上可接受的酸加成盐的实例包括与以下形成的那些盐:无机酸,如盐酸、氢溴酸、硫酸、硝酸、磷酸等;以及有机酸,如乙酸、三氟乙酸、丙酸、己酸、环戊烷丙酸、乙醇酸、丙酮酸、乳酸、草酸、马来酸、丙二酸、琥珀酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、3-(4-羟基苯甲酰基)苯甲酸、扁桃酸、甲磺酸、乙磺酸、1,2-乙二磺酸、2-羟基乙磺酸、苯磺酸、4-氯苯磺酸、2-萘磺酸、4-甲苯磺酸、樟脑磺酸、葡庚糖酸、4,4′-亚甲基双-(3-羟基-2-烯-1-甲酸)、3-苯基丙酸、三甲基乙酸、叔丁基乙酸、月桂基硫酸、葡萄糖酸、谷氨酸、羟基萘甲酸、水杨酸、硬脂酸、粘康酸、对甲苯磺酸等。
药学上可接受的碱加成盐的实例包括其中母体化合物中存在的酸性质子被金属离子置换而形成的那些盐,如钠盐、钾盐、锂盐、铵盐、钙盐、镁盐、铁盐、锌盐、铜盐、锰盐、铝盐等。具体的盐是铵盐、钾盐、钠盐、钙盐和镁盐。源自药学上可接受的有机无毒碱的盐包括但不限于,伯胺、仲胺和叔胺、取代的胺(包括天然存在的取代的胺)、环胺和碱性离子交换树脂的盐。有机碱的实例包括异丙胺、三甲胺、二乙胺、三乙胺、三丙胺、乙醇胺、2-二甲基氨基乙醇、2-二乙基氨基乙醇、二环己胺、赖氨酸、精氨酸、组氨酸、咖啡因、普鲁卡因、海巴明、胆碱、甜菜碱、乙二胺、葡糖胺、甲基葡糖胺、可可碱、嘌呤、哌嗪、哌啶、n-乙基哌啶、氨丁三醇、n-甲基葡糖胺、聚胺树脂等。示例性有机碱是异丙胺、二乙胺、乙醇胺、三甲胺、二环己基胺、胆碱和咖啡因。
术语“前药”是指例如通过在血液中水解而在体内转化(通常快速)以产生上式母体化合物的化合物。常见实例包括但不限于具有携带羧酸部分的活性形式的化合物的酯和酰胺形式。本发明的化合物的药学上可接受的酯的实例包括但不限于,烷基酯(例如具有约1个与约6个之间的碳),烷基是直链或支链(具有单个或多个支链)。可接受的酯还包括环烷基酯和芳基烷基酯,如但不限于苄酯。本发明的化合物的药学上可接受的酰胺的实例包括但不限于,伯酰胺以及仲烷基酰胺和叔烷基酰胺(例如具有约1个与约6个之间的碳)。可根据常规方法制备本发明的化合物的酰胺和酯。
如本领域的技术人员将了解,本发明的方法特别可用于治疗性或预防性治疗以局部缺血诱导的或局部缺血-再灌注诱导的心肌组织损伤为特征的病状。
因此,在一个相关方面,提供一种治疗性或预防性治疗哺乳动物中以局部缺血诱导的心肌组织损伤或局部缺血-再灌注诱导的心肌组织损伤为特征的病状的方法,所述方法包括向所述哺乳动物施用有效量的结构(i)的化合物:
或其药学上可接受的盐、溶剂化物或立体异构体,其中
(a)x是ch2;
y是ch2conh、(ch2)2conh、ch(ch3)conh、ch2、ch2co、ch2nhconh、ch2nhco、(ch2)2o、(ch2)2nh、(ch2)2nhconh、(ch2)2nhco、ch2csnh或c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(b)x是ch2;
y是ch2conh;以及
r1是ch3、h、ch2ch3、ch(ch3)2、c6h11、c6h5、2-噻吩基、c6h4-och3、c6h4-cl、c6h4-ch3、c6h4-f或ch2-c6h5;
或
(c)x是nhco或nhconh;
y是ch2;以及
r1是ch3;
或
(d)x是nh;
y是ch2conh;以及
r1是ch3;
或
(e)x是nhco或co;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是ch2co;
其中被r2和r3取代并与结构(i)的y键合的苯基被n-甲基哌嗪、n(ch3)-c6h4-br、nhch2-c6h4-br或o-c6h4-br置换;以及
r1是ch3;
或
(g)x是nhconh、nhco或nh;
结构(i)的y不存在,并且相反,被r2和r3取代的苯基直接键合至哒嗪酮环的n2;以及
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
或
(i)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(j)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(k)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(1)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(m)x是nh;
y是ch2conh;
r1是ch3;以及
c5另外地被coch3取代;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;以及
其中在足以使所述药剂诱导心肌细胞fpr1活化的时间内和条件下施用所述化合物。
在一个实施方案中,所述病状是冠状动脉粥样硬化、冠状动脉血凝块形成、心脏小动脉扩散性扩散性缩窄、冠状动脉手术夹紧、血管成形术、心肌梗塞、急性心肌梗塞、极低血压、创伤、出血或严重感染。
在另一个实施方案中,所述组织损伤包括心肌炎症、收缩功能丧失、细胞坏死、细胞凋亡、纤维化、心肌组织重塑、梗塞范围和/或左心室功能。
治疗的受试者通常是哺乳动物,如但不限于人、灵长类动物、家畜动物(例如,绵羊、牛、马、驴、猪)、伴侣动物(例如狗、猫)、实验室试验动物(例如小鼠、兔、大鼠、豚鼠、仓鼠)、捕获的野生动物(例如狐狸、鹿)。优选地,哺乳动物是人或灵长类动物。最优选地,哺乳动物是人。
“有效量”意指至少部分获得所需响应或延迟所治疗的特定病状的发作或抑制所述病状的进展或完全停止所述病状的发作或进展所需的量。所述量取决于待治疗的个体的健康和身体状况、待治疗的个体的分类组、所需防护程度、组合物的配方、医学情况的评估和其他相关因素而变化。预期所述量将属于可通过常规试验确定的相对宽泛范围。
本文中提及“治疗性”和“预防性”治疗应在其最广泛的背景下考虑。术语“治疗性”并不一定意味着治疗病状直到完全恢复。类似地,“预防性”并不一定意味着患者将不会发展一定程度的心脏组织损伤。因此,治疗性和预防性治疗包括改善心脏损伤的症状或预防心脏损伤或以其他方式降低发展心脏损伤的风险。术语“预防性”可被认为降低损伤的严重性或程度。
根据前述方面和实施方案,结构(i)的化合物可以是
其中
(a)x是ch2;
y是ch2conh;以及
r1是ch3;
或
(b)x以及被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(c)x以及被r4和r5取代的苯基不存在;
y是ch2conh;
r1是ch3;以及
哒嗪酮环的c4与c5之间的键是单键;
或
(d)x是nh2,并且被r4和r5取代的苯基不存在;
y是ch2conh;以及
r1是ch3;
或
(e)结构(i)的x不存在,并且相反,被r4和r5取代的苯基直接键合至哒嗪酮环的c4;
y是ch2conh;以及
r1是ch3;
或
(f)x是ch2;
y是c(r6r7)conh;
其中r6是h;
r7是ch3、c2h5、n-c3h7、i-c3h7、n-c4h9或c6h5;
或
r6是ch3;
r7是c2h5或ch3;以及
r1是ch3;
或
(g)x是ch2;
y是ch3conh
r1是ch3;
或
(h)x是ch2;
y是ch2conh或ch(ch3)conh;
被r4和r5取代并与结构(i)的x键合的苯基被3-呋喃基、3-噻吩基、2-噻吩基、1-萘基、2-吡啶基或3-吡啶基置换,其每一个任选地被卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基中的一个或多个取代;以及
r1是ch3;
其中r2、r3、r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基、硝基,或r2和r3和/或r4和r5是桥联的二烷氧基;
或
(i)x是ch2;
y是ch(ch3)conh
r1是ch3;
其中r2是氢且r3是对-溴,并且r4和r5独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基;
或
(j)x是ch2;
y是ch(ch3)conh
r1是ch3;
其中r2和r3独立地是氢、卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、烷基硫、氰基或硝基,并且r4是氢,且r5是间-甲氧基;
或
(k)x是ch2;
y是ch(ch3)conh
r1是ch3;
其中r2是氢且r3是对-溴,并且r4是氢且r5是间-甲氧基;
或其药学上可接受的盐、溶剂化物或立体异构体。
结构(i)的化合物包括但不限于:
以及其药学上可接受的盐、溶剂化物或立体异构体。
所公开的化合物可根据cilibrizzi等人在j.med.chem.2009,52,5044-5057;cilibrizzi等人,bioorg.med.chem.2012,20,3781-3792;crocetti等人,drugdev.res.2013,74,259-271;以及giovannoni等人,eur.j.med.chem.2013,65,512-528中的方法来制备。用于合成本发明的化合物的起始材料和中间体通常是可商购获得的或可通过常规有机化学方法制备。用于合成本发明的化合物及其中间体的合适方法描述于例如,j.march,advancedorganicchemistry,第3版(johnwiley&sons,newyork,1985);d.c.liotta和m.volmer,编辑,organicsynthesesreactionguide(johnwiley&sons,inc.,newyork,1991);r.c.larock,comprehensiveorganictransformations(vch,newyork,1989);h.o.house,modernsyntheticreactions第2版(w.a.benjamin,inc.,menlopark,1972);以及n.s.simpkins,编辑100modernreagents(theroyalsocietyofchemistry,london,1989)中。
本公开的结构(i)的化合物根据cilibrizzi等人,j.med.chem.2009,52,5044-5057;cilibrizzi等人,bioorg.med.chem.2012,20,3781-3792;crocetti等人,drugdev.res.2013,74,259-271;以及giovannoni等人,eur.j.med.chem.2013,65,512-528的程序来制备。
本发明的另一方面涉及如上文所定义的化合物在制造用于使哺乳动物中局部缺血诱导的心肌组织损伤的程度最小化的药物中的用途。
在另一方面,提供如上文所定义的化合物在制造用于使哺乳动物中局部缺血-再灌注诱导的心肌组织损伤的程度最小化的药物中的用途。
根据这些方面,在一个实施方案中,所述心肌组织损伤是细胞活力和收缩功能的丧失。
在另一方面,所述心肌组织损伤是心肌梗塞诱导的组织损伤,并且更具体地说,急性心肌梗塞诱导的组织损伤。
本发明的化合物可取决于施用方法以多种单位剂型施用。用于典型调节化合物的剂量是本领域的技术人员所熟知的。这类剂量通常是建议性质的,并且根据具体的治疗情形、患者或器官耐受性等进行调整。足以实现这一目的的药剂的量被定义为“治疗有效剂量”。对此用途有效的剂量方案和量,即“给药方案”,将取决于多种因素,包括心脏状况、损伤发作的预先存在或不存在、药物制剂和活性剂的浓度等。在计算器官的剂量方案时,还将考虑施用方式。剂量方案还必须考虑药物动力学,即化合物的吸收速率、生物利用度、代谢、清除率等。(参见例如,最新的remington’s;egleton和davis1997peptides18:1431-1439;langer1990science249:1527-1533)。
化合物可通过任何方便的方式施用,并且考虑在以取决于特定情况的量施用时显示治疗活性。变化取决于例如人或动物以及选择的调节剂。广泛范围的剂量可适用。考虑到患者,例如,可施用约0.1mg至约1mg化合物/天/千克体重。可调整剂量方案以提供最优治疗响应。例如,可每日、每周、每月或其他适合时间间隔施用若干分次剂量,或可如由情况的紧急性所指示,按比例减少剂量。
化合物可以方便的方式施用,如通过口服、静脉内(在水溶性情况下)、腹膜内、肌内、皮下、皮内或栓剂途径或植入(例如使用缓释分子)施用。所述化合物可以如上所述的药学上可接受的无毒盐的形式施用。如果活性成分将以片剂形式施用,则片剂可含有粘合剂,如黄芪胶、玉米淀粉或明胶;崩解剂,如海藻酸;以及润滑剂,如硬脂酸镁。
施用途径包括但不限于静脉内、腹膜内、皮下、皮内、肌内、鞘内、输注、口服、经iv滴注、贴剂和植入物。
根据这些方法,根据本发明定义的化合物可与一种或多种其他化合物或分子共同施用。“共同施用”是指在同一制剂中同时施用,或在两种不同制剂中通过相同或不同途径同时施用或通过相同或不同途径顺序施用。例如,主题化合物可与药剂一起施用以增强其作用。“顺序”施用是指在两种类型的分子的施用之间数秒、数分钟、数小时或数天的时间差。这些分子可以任何顺序施用。
适用于注射使用的药物形式包括无菌水溶液(在水溶性情况下)或分散体,以及用于临时制备无菌可注射溶液或分散体的无菌粉末剂,或者可呈适于局部施加的乳膏剂形式或其他形式。在制造和储存条件下,所述化合物必须稳定且必须在贮存中防止诸如细菌和真菌的微生物的污染作用。载体可以是含有以下各项的溶剂或分散介质:例如,水、乙醇、多元醇(例如,甘油、丙二醇和液体聚乙二醇等)、其适合的混合物、以及植物油。可例如通过使用包衣(如卵磷脂)、通过在分散情况下维持所需颗粒大小以及通过使用表面活性剂来维持适当的流动性。对微生物作用的防止可通过各种抗菌剂和抗真菌剂(例如对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸、硫柳汞等)来实现。在许多情况下,将优选包括等渗剂,例如糖或氯化钠。可注射组合物的延长吸收可通过在组合物中使用延迟吸收的拮抗剂(例如单硬脂酸铝和明胶)来实现。
无菌可注射溶液通过将所需量的活性化合物与以上列举的各种其他成分(根据需要)掺入适当溶剂中,随后过滤灭菌来制备。通常,通过将各种灭菌活性成分掺入含有基本分散介质和来自以上列举的所需其他成分的无菌媒介物中来制备混悬液。在用于制备无菌可注射溶液的无菌粉末的情况下,优选的制备方法是真空干燥和冷冻干燥技术,其产生活性成分加来自其先前无菌过滤溶液的任何其他所需成分的粉末。
当活性成分被适当地保护时,它们可例如与惰性稀释剂或与可吸收的可食用载体一起口服施用,或者其可被包封在硬壳或软壳明胶胶囊中,或者其可被压制成片剂,或者其可与饮食的食物一起直接掺入。对于口服治疗性施用,活性化合物可与赋形剂组合并且以可摄入片剂、口含片剂、锭剂、胶囊剂、酏剂、混悬剂、糖浆剂、糯米纸囊剂等形式使用。此类组合物和制剂应含有至少1重量%的活性化合物。当然,所述组合物和制剂的百分比可改变并且可合宜地在单位重量的约5%至约80%之间。此类治疗有用组合物中的活性化合物的量使得将获得适合的剂量。制备本发明的优选组合物或制剂,以使得口服剂量单位形式含有介于约0.1μg与2000mg之间的活性化合物。
片剂、锭剂、丸剂、胶囊剂等也可含有下文列出的组分:粘合剂,如树胶、阿拉伯树胶、玉米淀粉或明胶;赋形剂,如磷酸二钙;崩解拮抗剂,如玉米淀粉、马铃薯淀粉、海藻酸等;润滑剂,如硬脂酸镁;并且可加入甜味拮抗剂如蔗糖、乳糖或糖精,或调味拮抗剂如薄荷、冬青油或樱桃香精。当单位剂型是胶囊剂时,除上述类型的材料之外,它可含有液体载体。各种其他材料可作为包衣存在或以其他方式修饰剂量单位的物理形式。例如,可用虫胶、糖或两者包覆片剂、丸剂或胶囊剂。糖浆剂或酏剂可含有活性化合物、作为甜味拮抗剂的蔗糖、作为防腐剂的对羟基苯甲酸甲酯和对羟基苯甲酸丙酯、染料和调味剂,如樱桃或甜橙香精。当然,在制备任何单位剂型中使用的任何材料应当是药用纯的并且在所使用的量下基本上无毒。此外,活性化合物可掺入持续释放制剂和配方中。
本发明涉及g蛋白偶联受体(gpcr),且更具体地涉及甲酰肽受体(fpr)。更具体地,本发明涉及fpr1亚型。本文提出的本发明部分地基于fpr1亚型的激动作用,且具体地说fpr1erk/akt信号传导的选择性活化。已经确定对fpr1erk/akt信号传导具有选择性的调节剂提供开发与fpr1亚型相关的改进疗法的机会。
因此,本发明的另一方面涉及一种鉴别调节靶受体的活性的候选化合物的方法,所述方法包括:
a)使受体与候选化合物接触,
b)确定所述候选化合物与所述受体的结合;
c)选择活化所述受体的候选化合物;
其中所述靶受体是甲酰肽受体亚型fpr1;以及
所述候选化合物使所述fpr1亚型活化至比通过所述fpr1亚型的内源性配体或底物的活化更高的程度。
优选地,所述化合物选择性地活化erk/akt信号传导。
在其他实施方案中,候选化合物可以是协同激动剂、不可逆激动剂或反向激动剂。
在一些实施方案中,调节靶受体活性的候选化合物可以是靶受体的变构效应物。根据本发明的变构效应物可以是抑制性或负效应物,或者可以是刺激性或正效应物。通过特异性结合靶受体上的位点而不是天然底物结合位点来实现变构效应,并由此抑制或刺激受体的活性。
本领域的技术人员将了解,本文所述的本发明可进行除明确描述的那些以外的变化和修改。应了解本发明包括所有此类变化和修改。本发明还包括在本说明书中单独地或共同地提及或指示的所有步骤、特征、组合物以及化合物,以及所述步骤或特征中的任何两个或更多个的任何和所有组合。
实施例1:体外分析
材料和方法
surefireerk1/2磷酸化、akt1/2/3磷酸化和camp积聚试剂盒购自perkinelmerlifeandanalyticalscience(waltham,ma,usa)。fip-in中国仓鼠卵巢(cho)细胞、gateway质粒、潮霉素b、fluo-4m获自invitrogen(carlsbad,ca,usa)。杜尔贝科氏改良的伊格尔培养基(dulbecco’smodifiedeaglemedium)(dmem)和胎牛血清(fbs)购自gibco(gaithersburg,md,usa)和jrhbiosciences(lenexa,ks,usa)。ac2-26由chemileliva(chongqing,china)合成,并且bidi-001和bidi-002由anthembioscience(bangalore,india)合成。所有其他材料均购自sigma-aldrich(st.louis,usa),除非另外指示;并且具有分析等级或更高级别。
化合物:实施例中提及以下四种化合物。
动物:所有动物研究按照澳大利亚国家卫生和医学研究指南(nationalhealthandmedicalresearchofaustralianguidelines)进行,并且获得了阿尔弗雷德教育区医学研究(amrep)动物伦理委员会(alfredmedicalresearchofeducationprecinct(amrep)animalethicscommittee)的批准。将新生(1-2天龄)sprague-dawley大鼠(混合性别)饲养并圈养在amrep动物中心。
原代新生大鼠心室心肌细胞的分离:用于心肌细胞分离的所有材料均是组织培养级的。如前所述(irvine,jc等人,ajp,2013),通过连续酶促消化进行心肌细胞分离。将心肌细胞悬浮于补充有100u/ml青霉素、100μg/ml链霉素和10%fcs的无菌dmem中。将富含肌细胞的细胞悬浮液预先接种两次(在37℃下45分钟)以减少成纤维细胞污染。然后,在1%5-溴-2′-脱氧尿苷(brdu,以限制任何剩余成纤维细胞的增殖)存在下,将心肌细胞以0.5x106接种到12孔板上以用于乳酸脱氢酶(ldh)测定。
新生大鼠心肌细胞中的模拟缺血性-再灌注:在分离后至少48小时后,将心肌细胞的培养基改变为无血清低葡萄糖dmem。然后使用低氧室(qnainternational,melbourne,australia)使心肌细胞经受6小时缺氧(95%n2-5%co2),随后进行48小时复氧,全部在37℃下进行以模拟局部缺血-再灌注(i-r)。在初步研究中初始测定缺氧心肌细胞中ac2-26、bidi-001、bidi-002(超过0.3-3μm)的最佳浓度;对于所有试剂1μm被证明是最有效的,并且用于所有随后心肌细胞研究中。在新鲜无菌过滤的krebs缓冲液(118mmnacl、4.8mmkcl、1.2mmkh2po4、1.2mmmgso4、25mmnahco3、50mmedta、100mmcacl2)中用新鲜药物溶液替换所有常氧或缺血性心肌细胞的培养基。将激动剂溶解于dmem或krebs缓冲液中,并且存在持续缺氧和复氧的全部持续时间。在48小时复氧结束时,将培养上清液等分式样收集在冰上并储存在-80℃下以用于评估心肌细胞损伤。将样品在冰上解冻,并且根据制造商的说明书(promegainc,danecountry,wi,usa)使用cyto-tox-onetm均质膜完整性测定,经由释放到上清液中的ldh的水平确定细胞损伤评估。为了确认心肌细胞损伤的程度,还根据制造商的说明书,使用可商购获得的高灵敏度大鼠ctnielisa试剂盒(lifediagnosticinc,westchester,pa,usa)评价了上清液中ctn的水平。
cdnda构建体和稳定细胞系的产生:从invitrogen获得编码人甲酰肽受体(fpr)亚型hfpr1、hfpr2和hfpr3的pcdna3.1+中的cdna。根据制造商的说明书(invitrogeninc,carlsbad,ca,usa),将3种hfpr受体的序列通过pcr扩增并使用bp克隆酶(clonase)酶混合物克隆至gateway进入载体pdonr202tm中。随后使用lr克隆酶酶混合物(invitrogen)将pdonr201tm中的所有hfpr受体构建体转染至pef5/frt/v5/dest载体中。如前所述(valantc等人,pnas,2014),使用受体构建体(pef5/frt/v5/dest)来转染fip-incho细胞。使用500μg/ml潮霉素选择细胞以产生稳定表达每种受体构建体的细胞系。在含有5%co2的湿润气氛下,在37℃下在补充有10%fbs、16mmhepes和500μg/ml潮霉素的高葡萄糖dmem中维持和培养细胞。
在下文描述的各种测定中,将细胞以4x104个细胞/孔接种至96孔透明细胞培养板中,在37℃下在5%co2中在无血清培养基中培养过夜。
细胞内钙动员:如前所述(keov,p等人,2014)测定细胞内钙动员。简言之,将细胞使用ph7.4的hepes缓冲盐水(hbs)溶液[150mmnacl、2.2mmcacl2、2.6mmkcl、1.2mmmgcl2、10mmhepes、10mmd-葡萄糖、0.5%(w/v)牛血清白蛋白(bsa)和4mm丙磺舒]洗涤一次,然后在37℃下在5%co2中在黑暗中用fluo-4-am(1μm,在hbs/bsa/丙磺舒中)处理60分钟。然后将细胞进一步洗涤并置于hbs/bsa/丙磺舒溶液中,以用于使用flexstation3读板仪(moleculardevices,sunnyvale,ca)进行连接添加和测定。对于所有实验,将荧光信号的峰值变化归一化至100μmatp的细胞响应,其用作内部阳性对照。
细胞外信号调控的激酶1和2磷酸化(perk1/2)的测量:进行在thr202/tyr204时程实验下的初始erk1/2磷酸化以确定在通过每种激动剂刺激后erk1/2磷酸化最大的时间。将细胞以4x104个细胞/孔接种到透明96孔细胞培养板上。在6小时后,将细胞用pbs洗涤一次,且然后在无血清的dmem中孵育过夜,以使fbs刺激的磷酸化的erk1/2水平下降。然后将细胞与fmlp和bidi-002一起用激动剂刺激5分钟,并且对于bidi-001刺激7分钟(基于初始时程研究在此时间点的最优最大信号),并在37℃下在5%co2中孵育。10%(v/v)fbs用作阳性对照,还包括媒介物对照。通过除去研究药物并用100μl的surefire裂解缓冲液(tgrbiosciences)裂解细胞来终止反应。将细胞在surefire裂解缓冲液中裂解,并转移至384孔不透明的浅孔微孔板(proxiplate)。在低光条件下,制备alphascreen珠粒(受体和供体):surefire反应缓冲液的1∶120(v∶v)稀释液,将其分别与活化缓冲液以6∶1(v∶v)的比率混合。将板在37℃下在黑暗中孵育1小时,然后通过fusion-α读板仪(perkinelmerlifeandanalyticalsciences,fostercity,ca,usa)与标准alphascreen设定测量荧光信号。将数据归一化至由10%(v/v)fbs在同一时间点引发的最大响应。
如前所述(keov,p等人,2014),使用alphascreentmerk1/2surefiretm试剂盒测定受体介导的perk1/2水平。对于所有实验,10%fbs用作内部阳性对照以刺激perk1/2,并且将最大响应用于数据的归一化,而媒介物用作阴性对照。
akt1/2/3磷酸化和akt1的测量:将同一裂解物用于测量akt1/2/3磷酸化(ser473)和akt1(thr308)。将细胞在surefire裂解缓冲液中裂解,并转移至384孔不透明的浅孔微孔板。在低光条件下,制备alphascreen受体珠粒:surefire活化缓冲液:surefire反应缓冲液的1∶10∶40(v∶v∶v)稀释液,并将板在室温下在黑暗中孵育2小时。然后,制备alphascreen供体珠粒/稀释液的1∶20(v∶v)稀释液,并在室温下在黑暗中孵育另外2小时,然后通过使用fusion-a读板仪(perkinelmerlifeandanalyticalsciences)与标准alphascreen设定测量荧光信号。根据制造商的说明书,使用alphascreentmerk1/2surefiretm试剂盒测定akt1/2/3(ser473)和akt1(thr308)的受体介导的磷酸化。将数据归一化至由10%(v∶v)fbs在同一时间点引发的最大响应。
camp积聚:将cho-hfpr1、-hfpr2和-hfpr3稳定转染的细胞接种至96孔板中,并在37℃下在5%co2中培养过夜。将细胞用pbs洗涤并培养过夜。在测定前三十分钟,将培养基用刺激缓冲液[1.4mnacl、50mmkcl、8mmmgso4、2mmna2hpo4、4.4mmkh2po4、0.1%(w/v)bsa、5mmhepes、1.3mmcacl2、5.6mm葡萄糖)]和10mm咯利普兰(选择性磷酸二酯酶-4抑制剂)替换,并用毛喉素(3μm)处理,且在37℃下在5%co2中孵育30分钟。吸出培养基,并将camp进行乙醇萃取(100%冰冷)且干燥。将细胞在检测缓冲液(dh2o、0.3%tween20、5mmhepes、0.1%bsa)中裂解。将裂解物转移至384孔板,并且根据perkinelmercampalphascreen方案,将检测缓冲液/供体珠粒缀合的抗camp抗体和检测缓冲液:生物素化的camp:受体珠粒缀合的链霉亲和素的混合物添加至裂解物。在低光条件下,制备alphascreen受体珠粒:刺激缓冲液的1∶50(v∶v)稀释液,在384孔不透明的optiplate中制备检测缓冲液中的供体珠粒和50nm生物素化的camp的1∶150(v∶v)稀释液。将板在黑暗中在室温下孵育过夜,之后通过使用fusion-α读板仪(perkinelmerlifeandanalyticalsciences)测量荧光信号。将数据归一化至由10μm毛喉素在同一时间点引发的响应。
数据分析:使用graphpadprism6.0(graphpadsoftware,sandeigo,ca,usa)分析所有数据。在适当的情况下,拟合浓度-响应曲线以导出最大激动剂作用(emax)和配体效力估值(pec50),其中emax是系统(而不是激动剂)的最大可能响应,基础是在激动剂不存在下的基础响应水平,并且pec50是得到在emax与基础之间一半处的响应的激动剂浓度的负对数。
为了定量由每种激动剂跨各种途径([ca2+]i动员、perk1/2、akt1/2/3和camp积聚的抑制)介导的偏向激动作用,如前所述(stewartgd等人,2009)使用归一化至参考激动剂(在这种情况下,bidi-002)的转导比的操作模型重新拟合激动剂浓度-响应曲线。在这些条件下,如果测试激动剂和参考激动剂通过常见受体构象活化两种途径,则log[偏向性因子]应为0.0(或偏向性因子不同于1.0),不管途径之间的响应扩增的差异。相比之下,log[偏向性因子]与0.0的显著偏差(或偏向性因子不同于1.0)表明对于不同激动剂涉及不同构象。所有亲和力、效力、功效和协同性参数被估计为对数(christopoulos,1998)。除非另外说明,否则结果表示为平均值±s.e.。统计学分析通过学生t检验(student’sttest),或单因素方差分析、随后邓奈特氏比较(dunnett’scomparison)或图基事后检验(在适当的情况下)进行。p<0.05的值被认为是统计上显著的。
结果
fpr激动剂防止缺氧诱导的心肌细胞损伤:缺氧-复氧使ldh的释放显著增加约70%,而ctni(心肌细胞死亡的更灵敏的量度)与对照常氧心肌细胞相比几乎增加至三倍。当ac2-26从缺氧-复氧前5分钟存在并且在缺氧-复氧的持续时间内存在(1μm,图1a、1c)时,心肌细胞ldh和ctni释放显著降低。当仅在复氧期间存在时,ac2-26也显著减弱ctni水平的增加,ldh具有类似但不显著的趋势(分别图1b、1d,两者p=0.13)。
与ac2-26类似,小分子模拟物bidi-001和bidi-002当在再灌注期间存在时显著降低ctni(图1d)。用小分子处理的心肌细胞中的ctn和ldh水平与常氧细胞不再不同(图1a、1b、1c)。
bidi-001和bidi-002活化hfpr受体:为了评估转染的cho细胞中fpr激动剂(fmlp、bidi-001和bidi-002)的功能特性,使用稳定表达hfpr1、hfpr2和hfpr3的cho细胞系,并且监测细胞内ca2+动员(图2)、ekr1/2的磷酸化(图3)、akt1/2/3的磷酸化(图4)以及毛喉素刺激的camp积聚的抑制(图5)。所述激动剂没有一种在稳定表达hfpr3的cho细胞中介导响应(数据未示出),从而表明cho细胞具有最小基础hfpr表达。
由于已知fmlp通过两种gpcr(高亲和力fpr1和低亲和力fpr2)诱导ca2+通量,所以测试了cho细胞中具有过度表达的hfpr表达情况下bidi-001和bidi-002的作用。激动剂fmlp在具有纳摩尔范围内的pec的hfpr1-cho细胞和在具有微摩尔范围内的pec50的hfpr2-cho细胞中诱导浓度依赖性ca2+动员(表1)。这些结果证实了先前报道,即hfpr对fmlp具有高亲和力,而hfpr2对fmlp的亲和力低得多(prossnitz,e.r.和r.d.ye1997;ley.y.等人,1999)。fmlp在介导erk(图3,表2)和akt(图4,表3)的磷酸化方面也具有类似的对hfpr1的超过hfpr2的优先活化。
在hfpr1和hfpr2细胞两者中,bidi-001介导浓度依赖性ca2+动员(图2)以及erk1/2(图3,表2)和akt1/2/3(图4,表3)的磷酸化。此外,与hfpr2相比,bidi-001在活化hfpr1方面更有效,在微摩尔范围内高约3倍。相比之下,在hfpr1和hfpr2细胞两者中,bidi-002介导浓度依赖性响应,且对ca2+动员(图2)、erk(图3,表2)和akt(图4,表3)的磷酸化具有类似的效力(约100μm)。
还测定了通过camp途径进行的信号传导。图5a说明hfpr激动剂在分别抑制camp积聚hfpr1和hfpr2下方面的浓度-响应曲线。没有激动剂能够引起毛喉素刺激的响应的完全抑制;所有作用均小于50%最大抑制。令人感兴趣地,在低激动剂浓度下发生的fmlp和bidi-002对hfpr1中毛喉素刺激的camp积聚的抑制在较高浓度下逆转,从而产生明显钟形的曲线。可将数据拟合至允许测定抑制期的激动剂效力的经验模型。另一方面,bidi-001在hfpr1下以浓度依赖性方式引起camp积聚。此外,在hfpr2下测试的浓度范围内,fmlp、bidi-001和bidi-002引起camp响应的抑制。表4总结三种fpr激动剂在刺激抑制毛喉素刺激的camp方面的最大响应范围,以毛喉素的百分比表示。总之,与对于hfpr亚型具有有限选择性的bidi-001相比,bidi-002在刺激hfpr受体方面更有效。
bidi-001是偏向性激动剂:为了评估配体展示偏向性激动作用的潜力,将来自[ca2+]i动员、erk1/2磷酸化、akt1/2/3磷酸化和camp积聚的结果以相对于bidi-002的偏向性曲线(图6)的形式重新绘制,其显示与[ca2+]i动员的最小联结。bidi-001偏向远离[ca2+]i动员,从而表明它可能能够产生hfpr1和hfpr2的不同构象。将激动作用的操作模型应用于数据允许相对于bidi-002的偏好定量偏向程度,其中发现bidi-002对akt磷酸化、erk磷酸化和camp积聚具有大约20倍偏好。这些发现代表迄今为止在hfpr受体下鉴别的最大程度的偏向激动作用。
实施例2:体内分析
材料和方法
动物和手术:所有动物研究按照澳大利亚国家卫生和医学研究指南进行,并且获得了阿尔弗雷德教育区医学研究(amrep)动物伦理委员会的批准。
将雄性c57bl/6小鼠饲养并圈养在amrep动物中心中并维持在12小时亮/暗循环下。在麻醉后(i.p.,氯胺酮80mg/kg,赛拉嗪20mg/kg和阿托品1.2mg/kg),将小鼠随机分配至24小时、48小时和7天再灌注组,并如所描述(gaoxm,2011;jmcc,50;gaoxm,2000,jmcc)进行可逆左动脉降支(lad)结扎。在镇静后,对小鼠插管并用与氧混合的室内空气(潮气量0.25ml,150次呼吸/分钟)通气(harvard设备,ma,usa),并置于加热垫上。左胸廓切开术将经由跳动心脏所在的约第三肋间隙进行。
使用具有封闭两个释放环的活结的7-0丝缝线将lad可逆地结扎。区域性局部缺血通过结扎区域的浅色证实。然后将空气从胸腔排出,将腔闭合并恢复正常呼吸。在40-60分钟的局部缺血后,通过释放所述活结重新建立通过左冠状动脉的血流。假手术小鼠进行相同的手术程序,除了冠状动脉未结扎。安排了三个小鼠组群。使第1组群经受40分钟缺血性-24小时再灌注以用于评估心脏坏死。使第2组群经受60分钟闭塞与48小时再灌注,以用于定量心脏和全身炎症。使最后一组群经受7天再灌注,并评估早期心脏纤维化。
用研究药物处理:将动物随机分配至不同组,并在lad再灌注前5分钟施用所有处理。经由尾静脉静脉内(i.v.)注射肽ac2-26(1mgkg-1)(chemieliva,chongqing,china)及其媒介物(盐水中的10%二甲亚砜(dmso))。两者均由anthembioscience(bangalore,india)合成的fpr激动剂bidi-001(50mgkg-1,n=10)和bidi-002(50mgkg-1,n=7)及其媒介物对照(在盐水中含有0.8%tween20的10%dmso)作为悬浮液经由i.p.注射施用。初步研究表明,50mgkg-1i.p.导致在单次剂量后约8小时在微摩尔浓度范围内的两种化合物的血浆浓度(未公布的观察结果)。小鼠然后每天接受药物或媒介物注射,直到实验终点。两个不同的媒介物组反映不同的研究药物溶解度和施用途径。
样品收集:在研究期(24小时、48小时或7天)后,对梗塞或假手术小鼠进行安乐死,使用25g针和涂覆有肝素(500u/ml)的1ml注射器通过心脏穿刺收集血液。在24小时再灌注后从小鼠收集血浆,以评估心肌肌钙蛋白i(ctni)的水平;在48小时再灌注后从小鼠收集全血,以评估循环中的总白细胞和分类白细胞(wbc)数目。将肺、心房、左心室和右心室解剖,吸干并称重。将左心室(lv)在闭塞部位切成两半,其中将上部固定在中性缓冲液福尔马林(neutralbufferformalin)(nbf,thermofisherscientific,melbourne,australia)中,并且将顶部置于tissue-
心脏坏死的评估:用于评估心脏坏死的最佳时间点是缺血性损伤后24小时再灌注。根据制造商的说明书,使用可商购获得的高灵敏度大鼠ctnielisa试剂盒(lifediagnosticinc.,pennsylvania,usa)测定ctni的血浆水平。为了评价与风险区域(aar)、梗塞范围(is)和lv大小相关的梗塞范围,进行2,3,5-三苯基氯化四氮唑(ttc)染色。在24小时再灌注后,将lad在麻醉的小鼠中重新紧密闭塞,并将伊文思蓝(evansblue)染料(0.1ml,5%)作为团注注射至lv中。然后将心脏切除并在冷盐水中漂洗以除去过量染料。将lv分离,在-20℃下冷冻且然后横向切割成6至7个厚度1.0mm的切片。在37℃下将lv切片与1.5%ttc溶液一起孵育45分钟。伊文思蓝的存在指示灌注,而其不存在指示此区域缺乏灌注,而砖红色区域指示存活心肌,白色或淡黄色区域区别坏死组织。将切片固定在载玻片之间,并使用与数码相机(nikoncool-pix4500,tokyo,japan)耦合的olympus显微镜(leicawildm3b,heerbrugg,switzerland)数字采集图像。使用imagej分析程序(版本1.45s,nationalinstituteofhealth,usa)分析图像。盲法概述并定量非缺血区(蓝色区域)、风险区(aar,红色和白色或黄色区域)、梗塞区(白色或黄色区域)和总lv。梗塞范围被计算为梗塞区/风险区的百分比。
心脏炎症的评估:在60分钟局部缺血和48小时再灌注后将包埋于oct化合物中的lv组织以6μm切片以用于免疫荧光分析。将切片与4%多聚甲醛一起预孵育20分钟并且再与10%正常山羊血清一起预孵育30分钟。然后将切片在室温(rt)下与cd68+第一抗体或ly-6b.2第一抗体(1∶200,abdserotec,raleigh,usa)一起孵育1小时,然后与alexafluor546第二抗体(1∶200,invitrogen,carlsbad,usa)一起孵育30分钟,以分别检测心脏巨噬细胞和嗜中性粒细胞浸润。最后,将切片与0.001%hoechst33342(invitrogen,melbourne,australia)一起孵育30分钟,以引发细胞核染色。使用nikona1r共焦显微镜(nikoninstrumentsinc.,ny,usa)在20x放大倍数下拍摄单一图像,且然后使用nis-elementsar软件(4.10版用于windows,nikoninstrumentsinc.,ny,usa)将9x9个单一图像自动拼接在一起以形成每个完整lv切片。梗塞区域的图像在40x放大倍数下捕获,且然后使用fiji软件(1.48c版用于maxosx,nationalinstitutesofhealth,usa)分析所述拼接图像。为每个图像设定阈值,以能够最佳感测高于阈值的红色荧光强度,且然后软件自动定量这些红色荧光信号。
全身炎症的评估:将从经受60分钟局部缺血和48小时再灌注的小鼠收集的血液样品1∶20稀释到2%乙酸中以裂解红细胞用于使用血细胞计数器进行总wbc计数。使用hemacolor染色试剂盒(merckmillipore,melbourne,australia)对血液涂片进行染色,且然后使用olympusbiologicalchs显微镜(olympusinc.,tokyo,japan)在60x放大倍数下检查以用于手动计数每100个wbc的嗜中性粒细胞、淋巴细胞和单核细胞。
心脏组织学和纤维化的评估:由alffedpathologyservice(melbourne,australia)将在40分钟局部缺血和7天再灌注后固定在nbf中的lv心脏包埋在石蜡中,并用leica2135超薄切片机(leicamicrosystems,wetzlar,germany)以4μm切片。将切片用苏木精和曙红(h&e)染色以用于评估心肌形态,或用苦味酸天狼猩红(0.1%,fluka,bucks,switzerland;ph2)染色以用于评估心脏胶原沉积。在1.25x下拍摄图像以捕获完整lv。为了检查局部梗塞区域,使用显微镜(olympusbx61,olympusinc.,ontario,canada)和qcapturepro软件(5.1版用于windows,mediacyberneticinc.,maryland,usa)在20x下采集图像。基于细胞核的蓝色染色强度(1=最小,5=最大,由独立观察者分类),将h&e图像以1至5的标度按随机顺序盲法评分。对于来自48小时再灌注和7天再灌注研究的多组lv载片,使用image-proplus软件(mediacyberneticinc.,maryland,usa)定量测量苦味酸天狼猩红染色的区域(lv区域%)。
统计分析:使用graphpadprism6软件(graphpadprismsoftwareinc,lajolla,usa)进行图形建构和统计分析。数据呈现为平均值±平均值的标准误差(sem)。进行单因素方差分析(anova)与图基事后分析,其中n>3,并且p<0.05被认为是统计上显著的。
结果
i-r损伤后的全身和心脏特征:在再灌注开始后24小时、48小时和7天测定心脏损伤的延长,仅在48小时和7天再灌注小鼠群组中记录全身特征。
测量体重和器官重量以检查药物处理对经受心肌局部缺血-再灌注损伤或假手术的小鼠的全身和心脏特征的影响。尽管在两种媒介物(盐水中的10%dmso)i.v.对比盐水中的含有0.8%tween20的10%dmsoi.p.)之间不存在差异,但是将来自这两组的数据合并用于组分析。如表1中所示,在心脏缺血性损伤后48小时再灌注,在体重、心房重量或肺重量方面不存在差异,但当归一化至体重时,在媒介物、ac2-26和bidi-001处理的心脏中心脏重量显著增加。
对于梗塞心脏存在类似但不显著的趋势:在7天再灌注后,与假手术相比,媒介物情况下的体重比值明显(p=0.07),但在ac2-26处理的心脏中不再明显(p=0.97)。令人感兴趣地,施用bidi-001而不是ac2-26或bidi-002显著降低归一化至体重的总心脏重量和lv重量两者,这与bidi-001对i-r损伤后心脏重塑的保护作用一致。
ac2-26和bidi-001显著降低血浆心脏坏死:lad的闭塞和随后的再灌注产生在24小时时间点可靠测量到的小鼠lv中的明显损伤。大约60%的被伊文思蓝染色的lv,在风险区域(aar,图7b)下面,心室的这部分的30%-40%被梗塞(图7c)。在lad再灌注前立即施用肽ac2-26使梗塞范围显著减弱约25%。令人感兴趣地,小分子模拟物bidi-001使梗塞范围减少大约34%(而不是bidi-002,图7c)。血浆ctni水平是心肌细胞损伤的选择性标志物。与假手术相比,24小时再灌注后血浆ctni水平显著升高(图7d)。虽然在ir损伤后与假手术后相比,ctni水平在ac2-26处理的小鼠中仍然显著增加,但相对于媒介物处理的i-r损伤的小鼠,ac2-26降低ctni水平的非显著趋势是明显的(p=0.10,图7d)。令人兴奋的是,小分子模拟物bidi-001显著降低血浆中的ctni(在bidi-002情况下未观察到)。
ac2-26和bidi-001减弱心脏炎症:首次通过h&e染色的评分建立心肌i-r和处理对60分钟缺血性-48小时再灌注后心脏炎症的影响。与假手术组相比,心肌i-r组显示在所有组中在低倍(1.25x)下观察到的lv的局部区域中的h&e损伤评分,不管处理如何。这与此区域中的细胞浸润一致,如在较高放大倍数(20x,图8a)中所观察到。半定量分析揭示,与假手术相比,lv损伤评分在媒介物处理的i-r小鼠中明显增加;这种增加通过ac2-26处理显著抑制,但是相对于假手术,lv损伤在ac2-26处理的小鼠中仍然较大(图8b)。类似地,半定量分析还表明,媒介物、bidi-001和bidi-002处理的i-r小鼠中的lv损伤评分显著增加(图8c)。与媒介物处理的i-r小鼠相比,bidi-002(而不是bidi-001)显著减弱细胞浸润(图8c)。
为了进一步研究i-r损伤后心脏细胞浸润的性质,进行了巨噬细胞和嗜中性粒细胞的检测。lv切片中的嗜中性粒细胞的免疫荧光检测揭示,使用ly-6b.2的抗体,在20x和40x放大倍数(图9)下,与假手术相比,经受i-r的小鼠中的显著增加。与假手术相比,在媒介物和ac2-26处理的i-r小鼠中,i-r损伤引发每个切片嗜中性粒细胞信号的显著增加(图9b)。与盐水处理的i-r相比,施用ac2-26使嗜中性粒细胞数目减少大约35%(图9b)。与假手术相比,每个lv切片的ly-6b.2信号检测也在媒介物处理的i-r小鼠内显著增加(图9c)。bidi-001(而不是bidi-002)使ly-6b.2嗜中性粒细胞信号显著减弱几乎一半(图9c)
在媒介物处理的小鼠和ana-a1模拟物处理的小鼠两者中,与假手术相比,在60分钟局部缺血-48小时再灌注后,lv切片中的巨噬细胞数目在所有处理组中也增加(图10)。通过免疫荧光对巨噬细胞的cd68+检测表明,与假手术相比,每个lv切片的信号量在媒介物和ac2-26处理的i-r小鼠中显著增加,与媒介物处理的i-r小鼠相比,在ac2-26处理的i-r小鼠中观察到巨噬细胞信号的减少的非显著趋势(p=0.10)(图10b)。如图10c中所示,与假手术相比,在媒介物处理的小鼠中观察到cd68+信号的显著增加,其倾向于由bidi-001(p=0.11)和bidi-002(p=0.11,图10c)两者降低。
ac2-26和bidi-001倾向于减轻全身炎症:进行总wbc和分类wbc定量以研究anx-a1模拟物对全身炎症的作用(图11)。与假手术相比,总循环wbc的数目以及嗜中性粒细胞、淋巴细胞和单核细胞的分类wbc数目在媒介物处理的i-r小鼠中显著增加;但是在ac2-26处理的i-r小鼠中未观察到这一点(图11a、c、e、g)。ac2-26处理的小鼠未显著改变总wbc和分类wbc数目。bidi-001倾向于减少wbc数目(p=0.07)和淋巴细胞数目(p=0.08)(图11b、f),而bidi-002显著增加单核细胞的水平(图11h)。
ac2-26和bidi-001减弱心脏纤维化:对来自48小时再灌注研究和7天再灌注研究两者的lv切片进行苦味酸天狼猩红染色以检测胶原沉积。在盐水和ac2-26处理的小鼠中,在低倍(1.25x)下观察到的lv切片的局部区域中,48小时的再灌注未诱导任何明显的苦味酸天狼猩红染色增加(数据未示出),而7天再灌注引起所有经受心肌i-r损伤的小鼠中染色区域的明显增加(图12b)。定量分析揭示,与假手术相比,媒介物处理的i-r小鼠中红色染色的区域(lv区域%)明显增加;这种增加倾向于通过ac2-26处理得以抑制,但是以非显著的方式(p=0.08,图12c)。类似地,i-r诱导的心脏纤维化增加持续存在于媒介物和bidi-002处理的小鼠中,但在bidi-001处理的小鼠中不存在。
实施例3:进一步体内分析
动物:所有动物研究按照澳大利亚国家卫生和医学研究指南进行,并且获得了阿尔弗雷德教育区医学研究(amrep)动物伦理委员会的批准。将新生(1-2天龄)sprague-dawley大鼠(混合性别)和成年雄性c57bl/6小鼠饲养并圈养在amrep动物中心中并维持在12小时亮/暗循环下。所有试剂均购自sigma-aldrich(st.louis,usa),除非另外指示;并且是分析级或更高级的。
动物手术:将成年c57bl/6小鼠随机分配至体内心肌局部缺血-再灌注(i-r)损伤或假手术。麻醉小鼠(氯胺酮80mg/kg,赛拉嗪20mg/kg和阿托品1.2mg/kg,kxa,i.p.)进行可逆的左动脉降支(lad)结扎,如所描述(gao等人2000&gao等人2011)。在镇静后,对小鼠插管并用与氧混合的室内空气(潮气量0.25ml,150次呼吸/分钟)通气(harvard设备,ma,usa),并置于加热垫上。在跳动心脏所在的第三肋间隙周围进行左胸廓切开术。使用具有封闭两个释放环的活结的7-0丝缝线将lad可逆地结扎。区域性局部缺血通过结扎区域的浅色证实。然后将空气从胸腔排出,将腔闭合并恢复正常呼吸。随后在缺血期结束时通过释放活结重新建立通过左冠状动脉的血流(即再灌注)。研究了三个不同的小鼠群组。使第1群组和第2群组经受40分钟局部缺血与24小时或7天再灌注,这是分别用于评估体内心脏坏死和早期心脏重塑的最佳时间点。使第3群组经受60分钟lad闭塞与48小时再灌注,以用于定量心脏和全身炎症。假手术小鼠进行相同的手术,除了lad未结扎。将小鼠随机分配至在再灌注前立即施用fpr激动剂,cmpd17b或cmpd43(两者均50mg/kg/天i.p.,两者均由anthembioscience,bangalore,india合成)(3,4),或等体积的媒介物对照(在盐水中含有0.8%tween-20的10%dmso,i.p.)。基于先前针对cmpd43减轻耳部炎症所示的剂量(cilibrizzi等人.2009&burli等人2006)选择体内fpr激动剂剂量,其中初步研究表明对于剂量后数小时,两种化合物的血浆浓度在高亚微摩尔-低微摩尔范围内。bidi-001和bidi-002的化学结构和分子量在图21a中描绘。所有其他材料均购自sigma-aldrich(st.louis,usa),除非另外指示;并且是分析级或更高级的。对于所有三个群组,将梗塞或假手术小鼠在研究结束时在kxa麻醉下安乐死,并通过心脏穿刺收集肝素化血液。
体内心脏坏死的评估:用于评估心脏坏死的最佳时间点是缺血性损伤后24小时再灌注。根据制造商的说明书,首先使用可商购获得的高灵敏度小鼠ctnielisa试剂盒(lifediagnosticinc.,pennsylvania,usa)测定在此时间点心肌肌钙蛋白i(ctni)的血浆水平。为了进一步评价心脏坏死的程度,还使用2,3,5-三苯基氯化四氮唑(ttc)染色测定与风险区域(aar)相关的梗塞范围(is)。在24小时再灌注后,将lad在麻醉的小鼠中重新紧密闭塞,并将伊文思蓝染料(0.1ml,5%)作为团注注射至lv中。然后将心脏切除并在冷盐水中漂洗以除去过量染料。将lv分离,在-20℃下冷冻且然后横向切割成6至7个厚度1.0mm的切片。在37℃下将lv切片与1.5%ttc溶液一起孵育45分钟。伊文思蓝的存在指示灌注,而其不存在指示所述区域缺乏灌注。砖红色区域指示存活心肌,而白色或淡黄色区域区别坏死组织。将切片固定在载玻片之间,并使用与数码相机(nikoncool-pix4500,tokyo,japan)耦合的手术显微镜(leicawildm3b,heerbrugg,switzerland)数字采集图像。使用imagej分析程序(版本1.45s,nationalinstituteofhealth,usa)分析图像。盲法概述并定量非缺血区(蓝色区域)、aar区(红色和白色或黄色区域)、梗塞区(白色或黄色区域)和总lv。梗塞范围被计算为aar中梗塞区的百分比。
体内早期心脏重塑的评估:将在40分钟局部缺血和7天再灌注(用于评估心脏纤维化和凋亡的最佳时间点)后从第2群组的小鼠收集的lv组织固定在nbf中,通过alfredpathologyservice(melbourne,australia)包埋于石蜡中,并且用leica2135超薄切片机(leicamicrosystems,wetzlar,germany)以4μm切片。将切片用苦味酸天狼猩红(0.1%,fluka,bucks,switzerland;ph2)染色以评估心脏胶原沉积。在1.25x下拍摄图像以捕获完整lv。为了检查梗塞周围区域,使用显微镜(olympusbx61,olympusinc.,ontario,canada)和qcapturepro软件(5.1版用于windows,mediacyberneticinc.,maryland,usa)在20x下采集图像。对于来自7天再灌注研究的多组lv载片,使用image-proplus软件(mediacyberneticinc.,maryland,usa)定量测量苦味酸天狼猩红染色的区域(lv区域%)。使用cardiotac原位细胞凋亡检测试剂盒(trevigen,gaithersburg,md,usa)(huynh等人2012),还在石蜡包埋的心室切片中评估梗塞区域的凋亡水平。该方法通过使用末端脱氧核苷酸基转移酶酶检测细胞核dna断裂,所述酶将标记的核苷酸掺入到dna片段的游离3′-oh末端上。通过蓝色染色来区分阳性染色的凋亡细胞,而将阴性染色的细胞用核固红复染色。凋亡细胞被定量为非凋亡细胞的百分比,并且结果表示为假手术小鼠心脏中检测到的倍数水平。在最后一次剂量大约20小时后,由药物候选物优化莫纳什中心(monashcentrefordrugcandidateoptimization)经由超高效液相色谱法与正电喷雾电离检测,还测定在7天再灌注后第2群组小鼠中cmpd17b和cmpd43的血浆水平。
体内心脏炎症的评估:将来自第3群组中的小鼠(在60分钟局部缺血和48小时再灌注后,用于检测心脏白细胞浸润的最佳时机)的肺、心房、左心室和右心室解剖,吸干并称重。将lv在闭塞部位切成两半,并且将顶端半部置于tissue-
体内全身炎症的评估:还从第3群组中的小鼠(在60分钟局部缺血和48小时再灌注后)收集全血和血浆,以评估总和分类循环白细胞(wbc)数目及促炎性细胞因子il-1β的血浆水平。将血液样品1∶20稀释到2%乙酸中以裂解红细胞用于使用血细胞计数器进行总wbc计数。使用hemacolor染色试剂盒(merckmillipore,melbourne,australia)对血液涂片进行染色,且然后使用olympusbiologicalchs显微镜(olympusinc.,tokyo,japan)在60x放大倍数下检查以用于手动计数每100个wbc的嗜中性粒细胞、淋巴细胞和单核细胞。根据制造商的说明书,使用可商购获得的elisa试剂盒(elizakit,melbourne,vic,australia)测定血浆il-1β。原代大鼠心肌细胞和小鼠心脏成纤维细胞的分离和培养:用于细胞分离的所有材料均是组织培养级的。如先前所述(irvine等人2012&irvine等人3),通过连续酶促消化进行原代新生大鼠心脏心肌细胞(nrcm)和成年小鼠心脏成纤维细胞(amcf)的分离。在1%5-溴-2′-脱氧尿苷存在下将nrcm以1.3x105个细胞/cm2的密度接种到12孔组织培养板(用于模拟局部缺血-再灌注,经由缺氧-复氧,h-r)或60mm组织培养皿中(用于基因表达)。将amcf(通道#3)以9.5x104个细胞/cm2接种到60mm组织培养皿上,并且在研究之前使其生长至80%汇合。在无血清dmem中48小时后,使12孔组织板中的nrcm经受通过用无菌过滤的kreb缓冲液(118mmnacl、4.8mmkcl、1.2mmkh2po4、1.2mmmgso4、25mmnahco3、50mmedta、11.0mm葡萄糖、1.75mmcacl2)替换培养基诱导的模拟局部缺血,然后在缺氧(95%n2-5%co2,使用缺氧室,qnainternational,melbourne,australia)下在37℃下孵育6小时。在缺氧结束时,在fpr激动剂cmpd17b、cmpd43(两者均为1μm)或媒介物存在下用新鲜无菌过滤的kreb缓冲液替换kreb缓冲液,并且使nrcm在37℃下经受48小时复氧。在48小时复氧结束时,收集nrcm上清液以用于评估心肌细胞损伤。根据制造商的说明书,使用可商购获得的高灵敏度大鼠ctnielisa试剂盒(lifediagnosticinc.)测定ctni的心肌细胞释放。在fpr激动剂cmpd17b、cmpd43(两者均为1μm)或媒介物存在或不存在下,将60mm培养皿上分散的nrvm在37℃下在无血清dmem中孵育48小时。在48小时结束时,提取rna并进行实时pcr(如先前所述(huynh等人2012)),以测定大鼠fprs的相对基因表达(rfpr1、rfpr2和rfpr3,表示为从所使用的最大循环数40减去的阈值循环数ct)。注意,iuphar受体命名法为人甲酰肽受体(因此为fpr1等)使用大写字母,而首字母大写用于啮齿动物受体(因此fpr1等)(ye等人2009)。
在37℃下与促纤维化刺激物转化生长因子tgf-β(10ng/ml)一起孵育24小时之前,将amcf在无血清dmem中饥饿过夜。在tgf-β之前30分钟存在fpr激动剂cmpd17b、cmpd43(两者均为10μm)或媒介物。在24小时刺激结束时,如先前所述(5),提取rna并进行实时pcr,以测定ctgf和il-1β相对于管家基因18s的相对基因表达。在24小时刺激结束时还测定小鼠fprs的相对基因表达(mfpr1和mfpr2,表示为从所使用的最大循环数40减去的阈值循环数ct)。体外人fpr下的偏向信号传导的评估:(i)稳定转染的重组hfpr-cho细胞的产生和培养:cho细胞不表达天然fpr,且因此是用于稳定转染人fpr的理想细胞。在本研究中,fip-incho细胞和gateway质粒获自invitrogeninc(carlsbad,ca,usa)。根据制造商的说明书,将所有3种已知人fpr(hfpr1、hfpr2和hfpr3)的序列通过pcr扩增并使用bp克隆酶酶混合物克隆至gateway进入载体pdonr202tm中。随后使用lr克隆酶酶混合物(invitrogen)将hfprpdonr201tm构建体转染至pef5/frt/v5/dest载体中。如前所述(valant等人2014),使用受体构建体(pef5/frt/v5/dest)来转染fip-incho细胞。使用500μg/ml潮霉素选择细胞以产生稳定表达每种人受体构建体的细胞系。将细胞在37℃下在含有5%co2的湿润气氛下在补充有10%胎牛血清(fbs,来自jrhbiosciences,lenexa,ks,usa)和500μg/ml潮霉素b(invitrogeninc)的高葡萄糖杜尔贝科氏改良的伊格尔培养基(dmem)中维持并培养。将细胞以4x104个细胞/孔接种至96孔透明细胞培养板中,在37℃下在5%co2中在无血清培养基中培养过夜。对于细胞内ca2+动员、细胞外信号调控的激酶1和2磷酸化(perk1/2)和akt1/2/3磷酸化(ser473和thr308两者)以及camp积聚抑制中的每一个,进行了cmpd17b、cmpd43以及广泛研究的fpr-亚型-选择性激动剂n-甲酰基-met-leu-phe(fmlp,fpr1选择性激动剂,来自sigma-aldrich)和脂氧素-a4(lxa4,fpr2选择性激动剂,来自biomolinternational,plymouthmeeting,pa,usa)中的每种的浓度-响应测定。
(ii)细胞内ca2+动员:如先前所述(valant等人2014),在稳定转染的重组hfprcho细胞中测定细胞内ca2+动员。简言之,将细胞使用ph7.4的hepes缓冲盐水(hbs)溶液[150mmnacl、2.2mmcacl2、2.6mmkcl、1.2mmmgcl2、10mmhepes、10mmd-葡萄糖、0.5%(w(10)(10)(10):v)牛血清白蛋白(bsa)和4mm丙磺舒]洗涤一次,然后在37℃下在5%co2中在黑暗中用fluo-4-am(1μm,来自invitrogeninc,在hbs/bsa/丙磺舒中)处理60分钟(may等人2010)。然后将细胞进一步洗涤并置于hbs/bsa/丙磺舒溶液中,并且使用flexstation3读板仪(moleculardevices,sunnyvale,ca)进行测定。对于所有实验,将荧光信号的峰值变化归一化至100μmatp的细胞响应,其用作内部阳性对照。
(iii)perk1/2磷酸化的测量:surefireerk1/2磷酸化试剂盒购自perkinelmerlifeandanalyticalscience(waltham,ma,usa)。进行在thr202/tyr204下的erk1/2磷酸化的时程实验以确定在通过每种激动剂刺激后最大磷酸化的时间。将稳定转染的重组hfprcho细胞以4x104个细胞/孔接种到透明96孔细胞培养板上。在6小时后,将细胞用pbs洗涤一次,且然后在无血清的dmem中孵育过夜,以允许fbs刺激的磷酸化的erk1/2水平下降。然后基于初始时程研究将细胞用fmlp和cmpd43刺激5分钟,并且用cmpd17b刺激7分钟,并在37℃下在5%co2中孵育。10%(v∶v)fbs用作阳性对照,还包括媒介物对照。通过除去研究药物并用100μl的surefire裂解缓冲液(tgrbiosciences)裂解细胞来终止反应。然后将裂解物转移至384孔不透明浅孔微孔板。在低光条件下,制备alphascreen珠粒(受体和供体两者):surefire反应缓冲液的1∶120(v∶v)稀释液,将其分别与活化缓冲液以6∶1(v∶v)的比率混合。将板在37℃下在黑暗中孵育1小时,然后通过fusion-α读板仪(perkinelmerlifeandanalyticalsciences,fostercity,ca,usa)与标准alphascreen设定测量荧光信号。
(iv)akt1/2/3(ser473)和akt1(thr308)磷酸化的测量:surefireakt1/2/3磷酸化试剂盒也购自perkinelmerlifeandanalyticalscience。使用如上的相同裂解物,将细胞在surefire裂解缓冲液中裂解,并转移至384孔不透明的浅孔微孔板。在低光条件下,制备alphascreen受体珠粒:surefire活化缓冲液:surefire反应缓冲液的1∶10∶40(v∶v∶v)稀释液,并将板在室温下在黑暗中孵育2小时。然后,制备alphascreen供体珠粒/稀释液的1∶20(v∶v)稀释液,并在室温下在黑暗中再孵育2小时,然后通过使用fusion-α读板仪(perkinelmerlifeandanalyticalsciences)与标准alphascreen设定测量荧光信号。对于所有erk1/2和akt1/2/3磷酸化实验,10%fbs用作内部阳性对照以刺激perk1/2和pakt1/2/3,并且将最大响应用于数据的归一化,而媒介物用作阴性对照,如先前所述(may等人2010)。
(v)camp积聚:在刺激前三十分钟,将稳定转染的重组hfprcho细胞的培养基用刺激缓冲液[1.4mnacl、50mmkcl、8mmmgso4、2mmna2hpo4、4.4mmkh2po4、0.1%(w/v)bsa、5mmhepes、1.3mmcac12、5.6mm葡萄糖)]和10mm咯利普兰(选择性磷酸二酯酶-4抑制剂)替换,并用毛喉素(3μm)处理,且在37℃下在5%co2中孵育30分钟。吸出培养基,并将camp用100%冰冷乙醇萃取且干燥。将细胞在检测缓冲液(dh2o、0.3%tween20、5mmhepes、0.1%bsa)中裂解。使用也购自perkinelmerlifeandanalyticalscience的surefirecamp积聚试剂盒,将裂解物转移至384孔板,并且根据perkinelmercampalphascreen方案,将检测缓冲液/供体珠粒缀合的抗camp抗体和检测缓冲液:生物素化的camp:受体珠粒缀合的链霉亲和素的混合物添加至裂解物。在低光条件下,制备alphascreen受体珠粒:刺激缓冲液的1∶50(v∶v)稀释液,在384孔不透明的optiplate中制备检测缓冲液中的供体珠粒和50nm生物素化的camp的1∶150(v∶v)稀释液。将板在黑暗中在室温下孵育过夜,之后通过使用fusion-α读板仪(perkinelmerlifeandanalyticalsciences)测量荧光信号。将数据归一化至由10μm毛喉素在同一时间点引发的响应。
(vi)fpr激动剂效力和最大激动剂响应的导出:如先前所述,使用prism6.0(graphpad软件)进行计算机化非线性回归(kenakin等人2012&christopoulos等人2002)。所有亲和力、效力和功效被估计为对数。将通过每种激动剂跨五种途径(细胞内ca2+、perk1/2磷酸化、akt1/2/3(ser473)磷酸化、akt1(thr308)磷酸化和camp积聚的抑制)介导的浓度-响应曲线拟合以导出最大激动剂作用(emax)和配体效力估值(pec50),其中emax是系统对限定刺激物(而不是激动剂)的最大可能响应,而基础响应是在刺激物或激动剂不存在下,并且pec50是得到在emax与基础之间一半处的响应的激动剂浓度的负对数(图21b)。
(vii)偏向激动作用的定量:开发gpcr激动作用的定量框架允许对配体偏向性进行定量和统计学分析。使用这些数据,使用如先前所述的black-leff操作模型的扩展(图21b)(kenakin等人2012),通过首先将上述获得的多个信号传导浓度-响应曲线中的每个重新拟合来定量每种激动剂跨每个信号介导的偏向激动作用的潜力。首先对于从每个浓度-响应曲线引出的每个响应导出“转导比”(τ/ka,其中τ表示激动剂对每种gpcr构象状态的功效和ka亲和力)。然后通过将所述转导比归一化至参考配体(在这种情况下,cmpd43)和参考途径(其在此是akt1/2/3(thr308)磷酸化)来计算“偏向性因子”(δδ(τ/ka))。最后,将偏向性因子绘制在“偏向性网”上以可视化偏向激动作用的潜力。这种方法使得细胞背景对观察到的激动作用的潜在混杂影响无效,并且促进每种激动剂的定量交叉途径比较。在这些条件下,如果测试激动剂和参考激动剂通过常见受体构象活化两种途径,则log[偏向性因子]应为0.0(即偏向性因子1.0),不管途径之间的响应扩增的差异。相比之下,log[偏向性因子]远离0.0的显著偏差(即其中偏向性因子不同于1.0)表明对于不同激动剂,涉及不同构象。所有亲和力、效力、功效和协同性参数被估计为对数(kenakin等人2012)。
体内心脏损伤响应:如所描述(burli等人2006&cilibrizzi等人2009)合成小分子fpr激动剂cmpd17b和cmpd43(图21a)。在3个单独的c57bl/6小鼠群组中评估其对体内心肌i-r损伤的影响(参见在线增刊)(gao等人2011)。在再灌注前立即施用fpr-激动剂cmpd17b和cmpd43(如所述,50mg/kg/天i.p.)或媒介物(盐水中10%-dmso/0.8%tween20i.p.),随后剂量每24小时直到终点。在24小时再灌注(gao等人2011)后关于梗塞范围(2,3,5-三苯基氯化四氮唑[ttc])和血浆心肌肌钙蛋白i(ctni)两者评估心脏坏死。在48小时再灌注(gao等人2011)后在单独小鼠组群中跨心脏巨噬细胞(cd68+)和嗜中性粒细胞(ly6b.2+)含量、循环白细胞(wbc)和血浆白细胞介素il-1β测定心脏和全身炎症(在在线增刊中详述)。最后,在7天再灌注后,在单独组群中测定早期心脏重塑(凋亡、纤维化)(huynh等人2010)和血浆fpr激动剂水平。
体外心肌细胞损伤响应:如所描述(在线增刊)分离并培养新生大鼠心肌细胞和成年小鼠心肌成纤维细胞。使心肌细胞经受缺氧-复氧(h-r)以体外模拟i-r,其中cmpd17b、cmpd43或媒介物在复氧开始时加入。通过测量心肌细胞上清液中的心脏ctni来评估心肌细胞损伤。使心肌成纤维细胞经受24小时促纤维化转化生长因子(tgf)-β刺激,其中cmpd17b、cmpd43或媒介物在tgf-β之前30分钟加入。然后评估促纤维化结缔组织生长因子(ctgf)、促炎性il-1β、fpr1和fpr2的表达。
体外人fpr下的偏向信号传导的评估:关于表达重组人(而不是天然)fpr的稳定转染的中国仓鼠卵巢(cho)细胞的产生和培养以及由fpr激动剂cmpd17b和cmpd43介导的浓度-响应曲线的建构和分析的详细描述,参考在线增刊。将结果与广泛研究的fpr1选择性激动剂fmlp和fpr2选择性激动剂lxa4进行比较。对5种信号传导途径进行了检查,即perk1/2磷酸化、akt1/2/3(ser473)磷酸化、akt1(thr308)磷酸化、ca2+i的增加以及毛喉素诱导的camp积聚的抑制。将激动作用的操作模型拟合至每个浓度-响应曲线以估计在所评估的每种途径下每种激动剂的转导比(τ/ka),如所描述(图21b)(kenakin等人2012)。将转导比归一化至参考激动剂(cmpd43)和参考途径(akt1/2/3(thr308)磷酸化),从而产生偏向性因子并因此允许每种激动剂的定量交叉途径比较。
数据分析:使用graphpadprism6.0(graphpadsoftware,sandeigo,ca,usa)分析所有数据。除非另外说明,否则结果表示为平均值±sem。统计学分析通过学生t检验,或单因素方差分析、随后邓奈特氏或图基事后检验(在适当的情况下)进行。p<0.05的值被认为是统计上显著的。
结果
fpr激动剂cmpd17b减少体内心脏坏死:确定了fpr激动剂(50mg/kg,i.p.,就在24小时再灌注前施用)对小鼠心脏坏死的影响。在24小时后,在i-r组之间被鉴别为风险区域(aar,在伊文思蓝上大约60%)的lv没有差异(图13a、b)。在媒介物处理的i-r小鼠中的aar中,大约40%是梗塞的(图13a、c),并且血浆ctni水平显著升高(图13d)。证明了cmpd17b显著降低梗塞范围(降低约35%,图13c)和ctni水平(图13d)。相比之下,与媒介物处理的i-r小鼠相比,cmpd43缺乏心脏保护作用。
fpr激动剂cmpd17b减弱体内心脏和全身炎症:经由lv嗜中性粒细胞(抗ly-6b.2)和巨噬细胞(抗cd68)含量的免疫荧光检测评估了在48小时再灌注后i-r诱导的心脏炎症的程度。与媒介物处理的i-r小鼠相比,cmpd17b显著减弱i-r诱导的lv嗜中性粒细胞含量的增加,减弱几乎50%(图14a-b,补充图14a);在cmpd43处理的i-r小鼠中不存在这种心脏保护作用。与假手术相比,lv巨噬细胞含量在媒介物处理的i-r小鼠中也显著升高,但是这未通过任一种fpr-激动剂显著减弱(图14c-d,补充图14b)。
与假手术相比,循环总wbc在媒介物处理的小鼠中显著增加(图15a)。i-r诱导循环淋巴细胞(p=0.10)、嗜中性粒细胞(p=0.09)和促炎性il-1β(p=0.10)中的每一种的类似但不显著趋势,这与全身炎症一致(图15b-15e)。cmpd17b显著减弱循环淋巴细胞和il-1β的水平,而不影响循环单核细胞。此外,在cmpd17b处理的小鼠中,不存在i-r诱导的总wbc和嗜中性粒细胞的增加。相比之下,与假手术相比,cmpd43在48小时再灌注后显著增加循环单核细胞和淋巴细胞。观察结果一起表明cmpd17b(而不是cmpd43)减弱与再灌注损伤相关的早期炎症响应。
fpr激动剂cmpd17b减弱体内心脏重构:接下来,在7天再灌注后,测定fpr激动剂对i-r诱导的心脏细胞凋亡(通过tunel)和纤维化(通过苦味酸天狼猩红)的影响。心肌i-r显著增加心脏细胞凋亡(图16a、b)和胶原沉积(图16c、d、图22c)。cmpd17显著减弱梗塞区域中的lv凋亡。在cmpd17b处理的小鼠中,不存在i-r对此区域中的lv纤维化的影响。此外,cmp17b显著降低心脏和lv重量(归一化至体重,图16e、f,表7)。与媒介物处理的i-r小鼠相比,cmpd43再次缺乏这些心脏保护作用。
fpr激动剂cmpd17b在体外减少心肌细胞损伤响应:评估了cmp17b和cmpd43对原代心肌细胞和成纤维细胞中的细胞损伤响应的直接作用,所述两种细胞均表达天然啮齿动物fpr1和fpr2(图23)。使用h-r来在体外模拟i-r,证明了cmpd17b拯救了h-r后的心肌细胞ctni释放(图17a)。相对于对照,cmpd43处理的(而不是cmpd17b处理的)心肌成纤维细胞中对tgf-β的促纤维化ctgf响应仍然显著(图17b,p<0.01)。此外,tgf-β诱导的促炎性il-1β响应的趋势倾向于持续存在于cmpd43处理的(而不是cmpd17b处理的)心肌成纤维细胞中,但是这并不显著(图17c)。关于处理对细胞fpr1/2表达的影响,参考在线增刊(图23)。
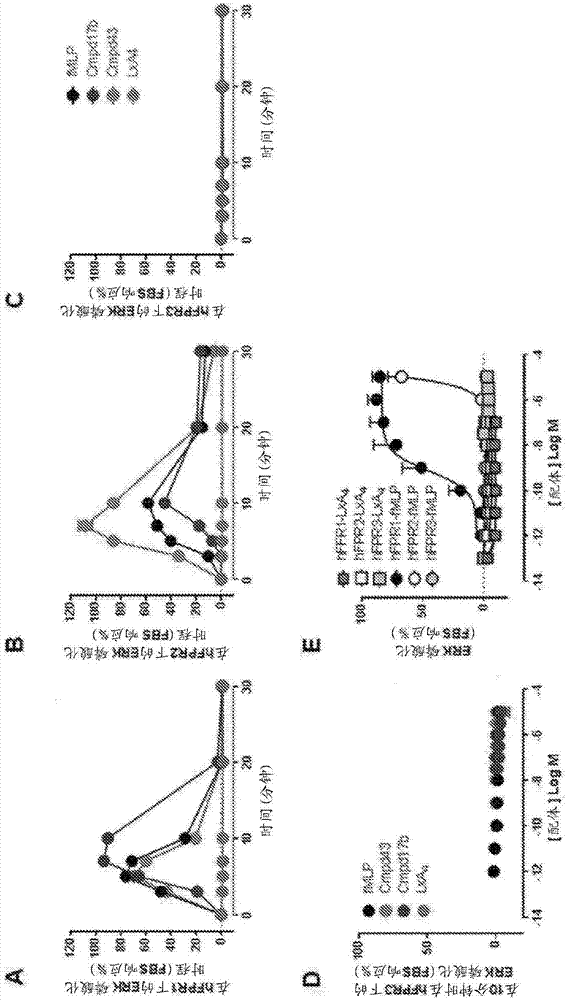
fpr激动剂cmpd17b和cmpd43活化人fpr1和fpr2:最后,在稳定表达人fpr1(hfpr1-cho)或人fpr2(hfpr2-cho)的flp-in-cho细胞中在体外评估了fpr激动剂cmpd17b和cmpd43的fpr1/fpr2信号传导指纹。在hfpr1-cho细胞(图18a-18d)和hfpr2-cho细胞(图19a-19d)中,cmpd17b和cmpd43两者均刺激关键心肌细胞存活途径erk1/2、akt1/2/3(thr308)和akt1/2/3(ser474)的浓度依赖性磷酸化,以及ca2+i的增加(对i-r后ca2+超负荷诱导的心肌细胞损失以及炎性细胞迁移来说是关键的)。在hfpr1-cho细胞(图18e)和hfpr2-cho细胞(图7e)中cmpd17b和cmpd43两者均抑制毛喉素刺激的camp积聚,这与fpr1/2与gi/o蛋白偶联、从而抑制腺苷酸环化酶一致。然而,在hfpr1-cho细胞中,cmpd43和fmlp在较高浓度下刺激camp积聚,从而产生双相曲线;因此,在抑制camp的浓度范围内导出ec50。在fpr1和fpr2两者下,对于所评估的每种信号传导途径,cmpd43表现出比cmpd17b更高的效力(表8)。在hfpr1-cho细胞中高亲和力fpr1选择性激动剂fmlp刺激ca2+i的浓度依赖性增加、camp积聚的抑制以及erk1/2、akt1/2/3(thr308)和akt1/2/3(ser474)的磷酸化,具有较高(纳摩尔)效力但与cmpd17b和cmpd43相似的最大值(图18a-e,表8)。推定fpr2选择性激动剂lxa4未能刺激任何hfpr亚型下游的erk磷酸化(图16)。在hfpr3-cho细胞中所测试的激动剂都未引发erk磷酸化(图24),从而证实所观察到的响应对相关hfpr1/hfpr2亚型的特异性。
cmpd17b是偏向性fpr激动剂:gpcr可在一系列构象状态之间异构化,每种信号传导响应均反映这些的特定子集。为了评估在给定gpcr下配体偏向性的潜力,必要的是将多种信号传导响应并入定量分析中;偏向性不能关于对单一响应的效力预测(kenakin等人,2012)。在本研究中,将激动作用操作模型拟合至五种信号传导途径中的每一种的浓度-响应曲线。这在每个信号传导终点产生每种激动剂的转导系数(τ/ka),其是提供对激动剂的总体功效的量度的配体功效(τ)和亲和力(ka)的复合值(kenakin等人2012)(图21b)。对于在hfpr1和hfpr2亚型下的每种下游信号,对于每种激动剂导出的偏向性因子在表8中示出。使偏向性因子可视化的“偏向性网”揭示,相对于cmpd43,在fpr1(图18f)和fpr2(图19f)两者下,cmpd17b显著偏向远离ca2+i,大约30倍。
体内i-r损伤后的全身和心脏特征:在表7中示出48小时和7天再灌注后测量的体重和器官重量。在缺血性再灌注48小时后,未观察到体重、心房重量或肺重量的差异。然而,当归一化至体重(bw)时,心脏湿重(hw)在媒介物和cmpd17b处理的i-r小鼠中显著增加。与假手术小鼠相比,心脏重量∶体重比(hw∶bw)存在的类似但不显著的趋势,在7天再灌注后在媒介物处理的i-r情况下是明显的(p=0.07,表7)。令人感兴趣地,施用cmpd17b(50mg/kg/天i.p.)显著降低总hw和归一化至bw的lv重量两者,这与cmpd17b对i-r后心脏重塑的保护作用一致。然而,这种心脏保护并不被在相同给药方案下的cmpd43共享,尽管在经受7天再灌注的小鼠中两种fpr小分子激动剂的血浆浓度在最后一次剂量后大约20小时超过0.1μm,其中血浆cmpd43水平在此时间点在微摩尔范围内。
体外nrcm和amcf中fprs的相对表达:如图23a中所示,常氧nrcm中fprs的相对表达是rfpr1>rfpr2>>rfpr3,其通过实时pcr评估。在48小时孵育后,cmpd17b未显著影响rfpr1(图23b)或rfpr2(图23c)的nrcm表达。相比之下,cmpd43倾向于降低rfpr2的nrcm表达(p=0.05),但对nrcmrfpr1表达没有影响。如图23d中所示,未处理的amcf中fprs的相对表达是mfpr2>mfpr1;未测定mfpr3。在24小时孵育后,tgf-β降低mfpr1(图23e)和mfpr2(图23f)两者的amcf表达。cmpd17b不显著影响mfpr1或mfpr2的amcf表达;相比之下cmpd43恢复mfpr1的amcf表达(图23)。
在体外hfpr下fpr激动剂作用的时程:lxa4的作用:为了阐明每种fpr激动剂对每种fpr亚型的最佳活化时间,进行了初步研究以确定响应于fmlp、cmpd17b、cmpd43和lxa4(各自10μm),erk1/2磷酸化的时程。如图24a和24b中所示,在hfpr1和hfpr2-cho细胞两者中,在用fmlp和cmpd43处理5分钟后观察到最大erk1/2磷酸化,而7分钟是cpmd17b的最大作用的时间点。在hfpr3下4种fpr激动剂都未引发作用(图24c和24d)。不管所研究的浓度,lxa4对任何hfpr亚型都无作用(图24e)。
实施例4
定量指纹图谱:bidi-001在体外在cho细胞中在fpr1下表现出远离ca2+i的偏向信号传导(但仍保留erk1/2&akt磷酸化完整)。重要的是,尽管具有较低cho细胞效力,但bidi-001(而不是cmpd43)在体外在分离的心肌细胞和在体内经受mi的完整心脏两者中均赋予心脏保护。这些结果表明偏向远离ca2+i(牵涉在mi后炎症和心肌细胞死亡两者中)可预测优异的体内心脏保护。化合物10394和10396的结果进一步证实了这一建议,其中仅10396在体外展示ca2+i偏向,并在体内赋予心脏保护(图25和表9)。
表1在稳定表达hfpr1受体或hfpr2受体的flpin-cho细胞中的效力(pec50)和最大激动剂[ca2+]i动员响应(emax)。数据点表示来自一式三份进行的3-7次实验的由atp引发的[ca2+]i动员的平均值±sem的百分比,n指示独立实验的数目。相对于fmlp,**p<0.01,****p<0.0001;相对于bidi-001,###p<0.001,####p<0.0001,单因素方差分析,然后邓奈特氏比较事后检验。
表2.在稳定表达hfpr1或hfpr2的flpin-cho细胞中的效力(pec50)和最大激动剂perk1/2响应(emax%)。数据点表示来自一式三份进行的4-6次实验的由10%fbs引发的erk1/2磷酸化的平均值±sem的百分比,n指示独立实验的数目。相对于fmlp,**p<0.01,****p<0.0001;相对于bidi-001,#p<0.05,单因素方差分析,然后图基事后检验。
表3.在稳定表达hfpr1或fpr2的flpin-cho细胞中的效力(pec50)以及最大激动剂pakt1/2/3(ser473)和pakt1/2/3(thr308)响应(emax%)。数据点表示来自4-5次实验的由10%fbs引发的akt1/2/3磷酸化的平均值±sem的百分比。相对于fmlp,**p<0.01,****p<0.0001;相对于bidi-001,#p<0.05,单因素方差分析,然后图基事后检验。
表4.在稳定表达hfpr1或hfpr2的flpin-cho细胞中的效力(pec50)和最大激动剂camp积聚抑制响应(emax)。数据点表示来自3-4次实验的对camp积聚的抑制的平均值±sem的百分比。相对于fmlp,**p<0.01,***p<0.001,单因素方差分析,然后图基事后检验。
表548小时心肌i-r和ac2-26、bidi-001、bidi-002处理对体重和器官重量的作用。相对于假手术,#p<0.05;相对于媒介物处理的小鼠,p<0.05。单因素方差分析与图基事后检验。数据表示为平均值±sem。
表67天心肌i-r和ac2-26、bidi-001、bidi-002处理对体重和器官重量的作用。相对于假手术,#p<0.05;相对于媒介物处理的小鼠,p<0.05。单因素方差分析与图基事后检验。数据表示为平均值±sem。
表7在i-r损伤后48小时和7天,在cmpd17b和cmpd43处理存在和不存在下,i-r对体重和器官重量的影响。
表8在稳定表达hfpr1或hfpr2的cho细胞中,对于erk1/2、akt1/2/3(thr308)、akt1/2/3(ser473)的磷酸化,细胞内ca2+动员和camp积聚抑制的fpr激动剂效力(pec50)、最大激动剂响应(emax)、转导系数(log(τ/ka))和“归一化的”转导系数(δlog(τ/ka))。数据表示来自一式三份进行的3-7次实验的由10%fbs(磷酸化)、atp(ca2+动员)或毛喉素诱导的camp积聚的抑制引发的响应的平均值±sem的百分比,n指示独立实验的数目。相对于fmlp,**p<0.01,****p<0.0001;相对于cmpd17b,###p<0.001,####p<0.0001;相对于细胞内ca2+动员,^^p<0.01,^^^p<0.001,单因素方差分析,然后图基事后检验。
表9bidi-001表现出在体外在cho细胞中在fpr1下远离ca2+i的偏向信号传导以及在体外在分离的心肌细胞和在体内经受mi的完整心脏两者中的心脏保护。nd=不可检测。
表10所测试的活性化合物。nd=不可检测。
参考文献
armstrongetal.,jama2007,297,43-51;
arumugam,t.v.,etal.,shock.2009,32(1),4-16;
s.m.bergeetal.,j.pharm.sci.,1977,66,1-19;
burlirw,xuh,zouxetal.bioorgmedchemlett2006;16:3713-8.
cha,j.,etal.,ann.thorac.surg.2008,85(5),1678-85;
chao,w.,am.j.physiol.heartcirc.physiol.2009,296(1),h1-12;
chen,g.y.andg.nunez,nat.rev.immunol.2010,10(12),826-37;
christopoulosa,kenakint.pharmacolrev2002;54:323-74.
cilibrizzietal.inj.med.chem.2009,52,5044-5057;
cilibrizzietal.,bioorg.med.chem.2012,20,3781-3792;
crocettietal.,drugdev.res.2013,74,259-271;
giovannonietal.,eur.j.med.chem.2013,65,512-528;
egletonanddavis,peptides,1997.18,1431-1439;
fleetwoodetal.,jimmunol.2007,178,5245-5252;
gao,x-metal.,j.mol.cell.cardiol.2000,32(9),1679-1686;
gaox-m,et.al.,j.mol.cell.cardiol.2011,50(6),991-999;
grangeretal.,circulation.2003,108,1184-1190;
house,h.o.,modernsyntheticreactions2ndedition(w.a.benjamin,inc.,menlopark,1972);
huynhk,kiriazish,duxjetal.diabetologia2012;55:1544-53.
hwangetal.,j.am.coll.cardiol.2001,38,1546-1553;
irvinejc,ganthaveev,lovejeetal.plosone2012;7:e44481.
irvine,j.c.etal.,am.j.physiol.2013,305(3),h365-377;
kenakint,watsonc,muniz-medinav,christopoulosa,novicks.acschemneurosci2012;3:193-203.
keov,p.etal.,j.biol.chem.2014,289(34),23817-37;
langer,science,1990,249,1527-1533;
larock,r.c.,comprehensiveorganictransformations(vch,newyork,1989);
ley.y.etal.,j.immunol.1999,163(12),6777-84;
leferandgranger,am.j.med.2000,109,315-323;
liotta,d.c.andvolmer,m.,eds,organicsynthesesreactionguide(johnwiley&sons,inc.,newyork,1991);
mahaffeyetal.circulation.2003,108,1176-1183;
march,j.,advancedorganicchemistry,3rdedition(johnwiley&sons,newyork,1985);
maylt,selftj,briddonsj,hillsj.molpharmacol2010;78:511-23.
mondenetal.,cardiovascres.2007,73,794-805;
prossnitz,e.r.,andr.d.ye.pharmacol.ther.1997,74(1),73-102;
remington’spharmaceuticalsciences,17thed.,mackpublishingcompany,easton,pa,1985;
simpkins,n.s.,ed.100modernreagents(theroyalsocietyofchemistry,london,1989);
stewartgdetal.,int.j.cancer2009,124(1),223-232;
sunetal.,circulation.2004,110,3221-3228;
suzukietal.,circulation.2001,104,i308-i303;
tavener,s.a.andp.kubes,am.j.physiol.heartcirc.physiol.2006,290(2),h800-6;timmersetal.,j.am.coll.cardiol.2009,53,501-510;
valantcetal.,pnas2014,111(12),4614-19;
ye,r.d.etal.,pharmacol.rev.2009,61(2),119-161.
- 还没有人留言评论。精彩留言会获得点赞!