前列腺特异性膜抗原(PSMA)的标记抑制剂,其作为显像剂和药剂用于治疗表达PSMA的癌症的用途
本发明一般涉及放射性药物的领域及其作为示踪剂、显像剂在核医学方面的用途和用于治疗表达psma的癌症的各种病况,特别是前列腺癌及其转移瘤。
相关技术
前列腺癌(pca)是美国和欧洲人口中的主要癌症。在西半球,至少一百万至两百万男性患有前列腺癌,并且估计55岁至85岁的每六个男性中就有一个会得患该病。在美国,每年有超过300000个前列腺癌的新病例。该疾病的死亡率仅次于肺癌。目前,具有高解剖分辨率的成像方法,例如计算机断层扫描(ct)、磁共振(mr)成像和超声,在前列腺癌的临床成像中占主导地位。据估计,目前全球每年用于手术、放射、药物治疗和微创治疗的费用为20亿美元。然而,目前没有有效的治疗复发性、转移性、雄激素非依赖性前列腺癌的方法。
目前在临床上实行了各种各样的实验性低分子量pca显像剂,包括放射性标记的胆碱类似物[18f]氟二氢睾酮([18f]fdht)、抗-1-氨基-3-[18f]氟环丁基-1-羧酸(抗[18f]f-facbc、[11c]乙酸酯和1-(2-脱氧-2-[18f]氟-l-阿拉伯呋喃糖基)-5-甲基尿嘧啶(-[18f]fmau)(scher,b.;等人,eurjnuclmedmolimaging2007,34,45-53;rinnab,l;等人,bjuint2007,100,786,793;reske,s.n.;等人,jnuclmed2006,47,1249-1254;zophel,k.;kotzerke,j.eurjnuclmedmolimaging2004,31,756-759;vees,h.;等人,bjuint2007,99,1415-1420;larson,s.m.;等人,jnuclmed2004,45,366-373;schuster,d.m.;等人,jnuclmed2007,48,56-63;tehrani,o.s.;等人,jnuclmed2007,48,1436-1441)。每一种都通过不同的机制起作用,并具有某些优点例如[11c]胆碱的尿排泄量低,还具有某些缺点例如发射正电子的放射性核素的物理半衰期短。
众所周知肿瘤可能表达与恶性表型有关的其独特蛋白质,或可能以比正常细胞更多的数量过度表达正常组成蛋白。肿瘤细胞表面上的独特蛋白的表达提供了通过探查肿瘤的表型特征、生化组成和活性来探测诊断和表征疾病的机会。选择性结合特定肿瘤细胞表面蛋白的放射性分子为在非侵入性条件下成像和治疗肿瘤提供了有吸引力的途径。一系列有前景的新的低分子量显像剂靶向前列腺特异性膜抗原(psma)(measer.c.等人,clincancerres.2008,14,3036-3043;foss,c.a.等人,clincancerres2005,11,4022-4028;pomper,m.g.等人,molimaging2002,1,96-101;zhou,j.等人,natrevdrugdiscov2005,4,015-1026;wo2013/022797)。
psma是跨膜的、750个氨基酸的ii型糖蛋白,其在pca表面有丰富且受限的表达,尤其是在雄激素非依赖性、晚期和转移性疾病中(schulke,n等人,procnatlacadsciusa2003,100,12590-12595)。后者很重要,因为几乎所有pca都随时间变成了雄激素非依赖型的。psma拥有用于治疗的有希望目标的标准(schulke,n.等人,proc.natl.acad.sci.美国,2003,100,12590-12595)。psma基因位于11号染色体的短臂上,并用作叶酸水解酶和神经肽酶。它具有相当于谷氨酸羧肽酶ii(gcpii)的神经肽酶功能,其称作“大脑psma”,并且可能通过将/v-乙酰天冬氨酸(naag)裂解为n-乙酰天冬氨酸(naa)和谷氨酸来调节谷氨酸能传递(nan,f.等人,jmedchem2000,43,772-774)。每个癌细胞最多有106个psma分子,进一步表明它是用基于放射性核素的技术进行成像和治疗的理想靶(tasch,j.等人,critrevimmunol2001,21,249-261)。
抗psma单克隆抗体(mab)7e11的放射免疫偶联物,其称为
无论是出于成像还是治疗目的,放射性药物对癌细胞的选择性靶向都具有挑战性。已知各种各样的放射性核素可用于放射成像或癌症放射治疗,所述放射性核素包括111in,90y,68ga,177lu,99mtc,123i和131i。最近,已显示与放射性核素-配体缀合物连接的含有谷氨酸-尿素-谷氨酸(gug)或谷氨酸-尿素-赖氨酸(gul)识别元件的一些化合物表现出对psma的高亲和力。在wo2015/055318中,描述了具有改善的肿瘤靶向性能和药代动力学的新的显像剂。这些化合物包含与癌细胞的细胞膜特异性结合的模体,其中所述模体包括前列腺特异性膜抗原(psma),其为上述的谷氨酸-尿素-赖氨酸模体。wo2015/055318中所描述的优选的分子还包括接头,所述接头通过酰胺键与作为螯合剂的dota的羧酸基相连接。已经显示这些化合物中的一些是用于前列腺肿瘤特异性靶向的有前景的试剂。用177lu(用于治疗目的)或68ga(用于诊断目的)标记化合物,并允许用于放射治疗目的的前列腺癌的可视化和靶向。
然而,仍然需要替代的或改善的配体,所述配体与psma相互作用并且带有合适的放射性核素,例如177lu或68ga,用于表达psma的癌症、特别是前列腺癌的检测、治疗和控制。
进一步地,应注意wo2015/055318中所述的化合物没有一种被描述与64cu和67cu形成稳定的配合物。然而,64cu/67cu对具有良好的性能,64cu非常适合长期pet成像,并且允许测定67cu标记的放射治疗剂的剂量。治疗性核素67cu是回旋加速器产生的,并且适用于gmp(良好生产规范)生产。它的半衰期允许使重复给药最佳化,仅需住院几天并降低了废物处理成本。由于t1/2=12.7小时,β+17.4%、e最大=0.656mev、β-39%、e最大=0.573mev)的特征,与68ga(t1/2=67.71分钟,88.9%β+)相比,64cu的量更大(wadastj,wongeh,weismangr,andersoncj.copperchelationchemistryanditsroleincopperradiopharmaceuticals.currpharmdes.2007;13:3-16.)。与68ga(67.71分钟)相比,64cu(12.7小时)充足的半衰期有利于在更后的时间点以及随后肿瘤轮廓增加时的成像(lewismr,wangm,axworthydb等人,invivoevaluationofpretargeted64cufortumorimagingandtherapy.jnuclmed.2003;44:1284-1292.21。desilvara,jains,learska等人,copper-64radiolabelingandbiologicalevaluationofbifunctionalchelatorsforradiopharmaceuticaldevelopment.nuclmedbiol.2012;39:1099-1104.)。这种放射性同位素的另一个主要优点是它可用于诊断以及用于治疗。因此,需要用铜放射性核素标记的psma配体。
进一步地,应注意在骨转移前列腺癌中,在用亲骨元素223racl2进行α射线治疗后观察到生存益处,其发射β的类似物89srcl2不能证明这一点(rubinig,nicolettia,rubinid,asabellaan.radiometabolictreatmentofbone-metastasizingcancer:from186reto223ra.cancerbiotherradiopharm.2014;29:1-11])。已经证明,对于患有神经内分泌肿瘤的患者,发射α的213bi-dotatoc可以克服对发射β的90y/177lu-dotatoc的耐药性[kratochwilc,gieselfl,bruchertseiferf,等人213bi-dotatocreceptor-targetedalpha-radionuclidetherapyinducesremissioninneuroendocrinetumorsrefractorytobetaradiation:afirst-in-humanexperience.eurjnuclmedmolimaging.2014nov;41(11):2106-19]。对基于α发射体的放射性核素治疗的兴趣持续增加。尽管出于研究目的而引起关注,但是213bi(t1/2=0.8小时)、212bi(t1/2=1.0小时)、149tb(t1/2=4.1小时)和211at(t1/2=7.2小时)的短物理半衰期对于可能的临床应用来说具有挑战性。在另一方面,如果大范围应用如用于流行病学重要肿瘤的治疗上,长半衰期α发射体例如227th(t1/2=18.7天)可能在环境中积累,并造成与废物处理有关的问题,而其长半衰期可能是应对全长抗体的缓慢药代动力学所必需的。强烈的辐射和相当短的路径长度是α发射体的主要特性。然而,只有几种适用于临床常规的具有合适半衰期的α发射体,例如212pb。
因此,仍然需要与α发射放射性核素形成合适的配合物的psma配体。
因此,本发明的总体目标是开发改善的配体,所述配体与psma相互作用并且带有合适的放射性核素,这为检测、治疗和管理表达psma的癌症、特别是前列腺癌提供了有利的选择。
技术实现要素:
所述目标的解决方案通过提供权利要求书中描述的实施方案来达成。发明人发现新的化合物,其是有用的和有利的放射性药物,并且可作为示踪剂、显像剂用于核医学和用于治疗表达psma的癌症,特别是前列腺癌的各种病况。在下文中更详细地描述这些化合物:
特别地,本发明涉及式(1)的化合物
或药学上可接受的盐或其溶剂化物,其中y3是o或s,其中s、t、u和w彼此独立地为0或1,其中i是1至3的整数,其中j是3至5的整数,其中z1、z2和z3彼此独立地选自-co2h、-so2h、-so3h、-oso3h和-opo3h2,r1是–ch3或h,优选h,x选自任选经取代的烷基芳基、芳基、烷基杂芳基和杂芳基,y1和y2彼此独立地选自任选经取代的芳基、烷芳基、环烷基、杂环烷基、杂芳基和烷基杂芳基,并且其中a是具有选自(ia)、(ib)和(ic)的结构的螯合剂残基,
其中r2、r3、r4和r5彼此独立地选自h、-ch2-cooh和–ch2-c(=o)-nh2或其中r2和r4形成-(ch2)n-桥,n为1至3的整数,其中n优选为2,其中r、v和q彼此独立地为0或1,条件是在u和w为0的情况下,q和v为0,并且其中
(a)u和w为1,或
(b)u为0和w为1,并且其中a选自(ia)或(ib),或
(c)a不是
进一步地,本发明涉及配合物,其包含
(a)放射性核素,和
(b)如上或如下所述的化合物,或其盐、溶剂化物、代谢物或前药。
进一步地,本发明涉及药物组合物,其包含如上或如下所述的化合物,或如上或如下所述的配合物。进一步地,本发明涉及如上或如下所述的化合物,或如上或如下所述的配合物,或如上或如下所述的药物组合物,其用于治疗、改善或预防表达psma的癌症和/或其转移瘤,特别是前列腺癌和/或其转移瘤。进一步地,本发明涉及如上或如下所述的化合物,或如上或如下所述的配合物,或如上或如下所述的药物组合物,其用于诊断。另外,本发明涉及如上或如下所述的化合物,或如上或如下所述的配合物,或如上或如下所述的药物组合物,其用于诊断癌症,特别是表达psma的肿瘤和/或其转移瘤。
在本发明的含义内使用的术语“表达psma的癌症和/或其转移瘤”涉及其癌细胞表达前列腺特异性膜抗原(psma)的任何癌症及其各自的转移瘤。优选地,可以根据本发明进行治疗的癌症(或癌细胞)选自前列腺癌、常规肾细胞癌、膀胱移行细胞癌、睾丸-胚胎癌、神经内分泌癌、结肠癌、脑肿瘤和乳腺癌。在本发明的特定优选的方面,所述表达psma的癌症是前列腺癌或乳腺癌,特别是前列腺癌。
如上所述,本发明的化合物或包含在配合物中的化合物具有以下结构:
应当理解化合物或包含在配合物中的化合物可以是阴离子形式的,或式(1)的化合物的盐形式的。
因此,本发明还涉及通式(1)的化合物或包含在配合物中的化合物的盐,特别是药学上可接受的盐。本发明还涉及这些化合物的溶剂化物,包括盐及其活性代谢物,以及在适当的情况下,其互变异构体,包括前药制剂。
“药学上可接受的盐”是本发明的化合物的药学上可接受的有机酸或无机酸或碱式盐。代表性的药学上可接受的盐包括例如碱金属盐、碱土金属盐、铵盐、水溶性和水不溶性盐,例如乙酸盐、碳酸盐、氯化物、葡萄糖酸盐、谷氨酸盐、乳酸盐、月桂酸盐、苹果酸盐或酒石酸盐。
术语“前药”是指药物的前体,其为化合物,一旦施用至患者则必须通过代谢过程经历化学转化,然后变成活性药剂。根据式(1)的说明性的化合物前药为酯和酰胺,优选烷基酯或脂肪酸酯。这里的前药制剂包括所有通过简单的转化形成的物质,所述转化包括酶促、代谢或以任何其它方式的水解、氧化或还原。合适的前药含有例如通式(1)的物质,其通过可酶切的接头(例如氨基甲酸酯、磷酸酯、n-糖苷或二硫化物基团)与改善溶解的物质(例如四甘醇、糖类、甲酸或葡糖醛酸等)连接。这种根据本发明的化合物的前药可以施用至患者,并且这种前药可以转化为通式(1)的物质,以获得期望的药理作用。
通式(1)的一些化合物可以以立体异构混合物的形式包括在内,例如外消旋混合物和/或顺式/反式异构体的混合物,或者作为单一对映异构体、非对映异构体和/或特定的顺式/反式异构体,包括其所有可能的混合物。
根据本发明,所有手性c原子都应该具有d构型和/或l构型;一种化合物内的组合也是可能的,即手性c原子中的一些可能是d构型而其他的可能是l构型。更优选地,化合物中存在的氨基酸残基具有l构型。
获得的化合物可以任选地通过已知的方法(例如,allinger,n.l.undelliele.l.in,,topicsinstereochemistry"vol.6,wileyinterscience,1971)在其对映异构体和/或非对映异构体中进行分离。对映体分离的一种可能的方法是使用色谱法。
尿素主链:
化合物(1)包含尿素构建单元(1a)。在化合物(1)的这种尿素构建单元(1a)中
z1、z2和z3彼此独立地选自co2h、-so2h、-so3h、-oso3h和-opo3h2,更优选z1、z2和z3中至少一种为-co2h,更优选z1、z2和z3均为-co2h。
应当理解,构建单元(1a)可以存在于任何立体异构形式中,然而,优选(1a)具有结构(1aa):
因此,优选本发明的化合物和包含在本发明的配合物中的化合物具有结构
整数i和整数j如上所述。
优选i为2。因此,本发明还涉及式(1),优选式(1a)的化合物,并且涉及包含在本发明的配合物中的化合物,其中i为2。
优选j为4。因此,本发明还涉及式(1),优选式(1a)的化合物,并且涉及包含在本发明的配合物中的化合物,其中j为4。
r1优选为h。
因此,尿素构建单元(1aa)最优选具有结构(1aa_1)
残基x和构建单元(1b):
如果s是1,式(1)的化合物包括构建单元(1b)
如上所述,在该构建单元中,x优选包括选自萘基、苯基、联苯基、吲哚基和苯并噻唑基的残基。优选x为萘基、烷基-萘基、苯基、苄基、联苯基、烷基-联苯基、吲哚基、烷基-吲哚基、苯并噻唑基或烷基-苯并噻唑基。
在本发明的含义中,术语萘基、苯基、联苯基、吲哚基和苯并噻唑基包括进一步被一种或多于一种合适的取代基取代的基团。在本发明的上下文中使用的术语“经取代的”优选指在任意位置被一种或多于一种取代基,优选1个、2个、3个、4个、5个或6个取代基,更优选1个、2个或3个取代基取代的基团。如果存在两种或多于两种取代基,每种取代基可以相同或可以与至少一种其它取代基不同。优选地,该基团是未经取代的。
在本发明的含义中,术语“烷基”涉及非支化烷基残基和支化烷基残基。该术语还包括进一步被一种或多于一种合适的取代基取代的烷基。在本发明的上下文中使用的术语“经取代的烷基”优选指在任意位置被一种或多于一种取代基,优选1个、2个、3个、4个、5个或6个取代基,更优选1个、2个或3个取代基取代的烷基。
更优选地,残基x选自:
其中这些基团可以被适当地取代。优选地,这些基团是未经取代的。
最优选地,x如果存在的话,x为
因此,构建单元(1b)如果存在的话,优选具有以下结构:
基团y1和构建单元(1c):
如果t是1,式(1)的化合物包括构建单元(1c)
如所述的,y1选自芳基、烷芳基、环烷基、杂环烷基、杂芳基和烷基杂芳基。
本发明的上下文中使用的术语“芳基”是指任选经取代的芳基,即特别是任选经取代的5元芳香环或6元芳香环,和经取代或未经取代的多环芳香基团(芳基),例如三环芳基或二环芳基。可提及任选经取代的苯基或萘基作为实例。多环芳香基团还可以含有非芳香环。
本发明的上下文中使用的术语“杂芳基”指任选经取代的杂芳基,即特别是任选经取代的5元芳香环或6元芳香环,和经取代或未经取代的多环芳香基团,例如三环或二环芳基,其在环系中含有一种或多于一种,例如1个至4个,例如1个、2个、3个或4个杂原子。如果环系中存在多于一种杂原子,则存在的至少两种杂原子可以相同或不同。合适的杂芳基是技术人员已知的。可以提及以下任选经取代的杂芳基残基作为非限制性实例:苯并二
本发明的含义内使用的术语“烷基芳基”或“烷基杂芳基”是指其中芳基或杂芳基通过烷基与构建单元各自剩余的部分连接的基团。因此,在x是例如c主链的情况下,本情况下的“烷基芳基”是指-烷基-芳基基团,“烷基杂芳基”是指-烷基-杂烷基基团。在y1的情况下,芳基或杂芳基通过烷基来与羰基连接,即因此在此情况下“烷基芳基”是指-烷基-芳基-基团,“烷基杂芳基”是指-烷基-杂芳基-基团。在y2的情况下,芳基或杂芳基通过烷基来与nh基团连接,即因此在此情况下,“烷基芳基”是指-烷基-芳基-基团,“烷基杂芳基”是指-烷基-杂芳基-基团。
在本发明的上下文中,术语“环烷基”是指任选经取代的环烷基残基,其中它们可以是单环基团或多环基团。可以提及任选经取代的环己基作为环烷基残基的优选实例。
本发明的上下文中使用的术语“杂环烷基”是指任选经取代的环烷基残基,其在环中具有至少一个杂原子,例如o、n或s,其中它们可以是单环基团或多环基团。
本发明的上下文中使用的术语“经取代的环烷基残基”或“环杂烷基”是指环烷基残基或环杂烷基残基,其中至少一个h被合适的取代基所取代。
优选地,y1是环烷基或杂环烷基,更优选环烷基,更优选
因此,构建单元(1c)如果存在的话,优选具有以下结构:
应当理解,这种结构包括任何可能的立体异构体,例如顺式/反式异构体。
优选地,该基团
作为反式异构体存在。因此,构建单元(1c)如果存在的话,优选具有以下结构:
基团y3
如上所述,y3优选为o或s。
放射性核苷酸
根据本发明的化合物是被用作放射显像剂还是放射性药物,不同的放射性核素与螯合剂络合。
说明性的放射性核素包括例如89zr、44sc、111in、90y、66ga、67ga、68ga、177lu、99mtc、60cu、61cu、62cu、64cu、66cu、67cu、149tb、152tb、153sm、155tb、161tb,153gd、155gd、157gd、213bi、225ac、230u、223ra、165er、fe的放射性核素(例如52fe和59fe)和pb的放射性核素(例如203pb和212pb、211pb、213pb、214pb、209pb、198pb、197pb)。
优选地,放射性核素选自111in、90y、68ga、177lu、153gd、155gd、213bi、225ac、60cu、61cu、62cu、64cu、66cu、67cu、fe的放射性核素(例如52fe和59fe)和pb的放射性核素(例如203pb和212pb、211pb、213pb、214pb、209pb、198pb、197pb)。
放射性核素pb更优选为203pb和212pb。
放射性核素cu更优选为64cu和67cu。
根据本发明的化合物的配合物可能含有一种或多于一种放射性核素,优选一种放射性核素。这些放射性核素优选适合用作放射显像剂或用作治疗增殖细胞,例如表达psma的癌细胞,特别是表达psma的前列腺癌细胞的疗法。根据本发明,它们被称为“金属配合物”或“放射性药物”。
优选的成像方法是正电子发射断层显像(pet)或单光子发射计算机断层显像(spect)。
实施方案(a)
根据本发明优选的实施方案,u和w为1。根据此实施方案,因此本发明的化合物具有以下结构。
在这种情况下,y3最优选为s。因此,本发明的化合物更优选具有以下结构。
如上所述,y2选自芳基、烷芳基、环烷基、杂环烷基、杂芳基和烷基杂芳基。更优选地,y2是芳基,更优选地,y2包含任选经取代的苯环,甚至更优选y2为
其中r6、r7、r8和r9彼此独立,为h或烷基、烯基、炔基、烷氧基、烷酰氧基、芳基、杂芳基、卤素、羟基、巯基、腈、胺,或在每种情况下任选取代的烷基、烯基、炔基、烷氧基、烷硫基、烷酰氧基(alkanoyloxy)、环烷基、苄氧基或芳基,更优选r6、r7、r8和r9彼此独立,为烷基或h,更优选r6、r7、r8和r9为h。
因此,本发明的化合物优选包括以下结构。
出人意料地发现利用这种化合物,可以优化与psma的相互作用。利用根据本发明的化合物,可以达到改善的肿瘤-非靶组织的积累和组织的值并且可以在非靶组织中获得改善的分布模式。
而且,根据本发明施用的螯合剂允许不能用于靶向表达psma的肿瘤的放射性核素与实际已知的示踪剂稳定结合。
如上所述,通常,螯合剂选自(ia)、(ib)和(ic)。
在实施方案a的情况下,如上和如下所述,整数r优选为0。更优选地,a是螯合剂,其选自
因此,本发明还涉及如上和如下所述的化合物和包含所述化合物的配合物,所述化合物具有以下结构。
更优选地,具有以下结构:
其中a是螯合剂,其选自
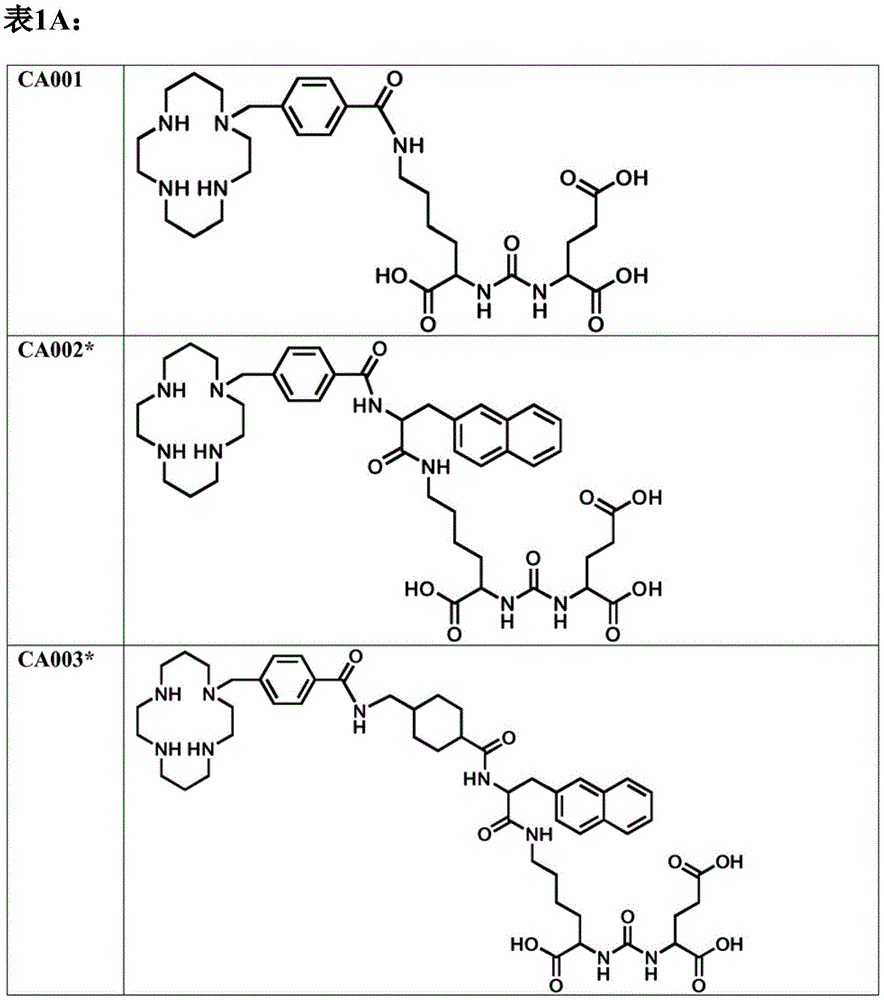
根据本实施方案的优选化合物的结构选自ca007、ca008*、ca009*、ca029*和ca030*(见表1a),其中更优选化合物ca007、ca008*和ca009*。应当理解,如果未指定表1a中相应所示结构中的立构中心,那么这意味着所有相应立体异构体都应以单独形式以及相应立体异构体的混合物形式包括在内。因此,化合物ca008*、ca009*、ca029*和ca030*可以以单一立体异构体或立体异构体的混合物存在。更优选地,根据本实施方案,化合物的结构选自ca007、ca008、ca009、ca029和ca030(见表1b),其中特别优选化合物ca007、ca008和ca009。
已经出人意料地表明,这些化合物显示出对psma的高结合亲和力并且被有效地内化。
实施方案(b)
根据本发明另一个优选的实施方案,u为0并且w为1。根据此实施方案,因此本发明的化合物具有以下结构。
根据该实施方案,a具有结构(ia)或结构(ib)。
如上所述,y2选自芳基、烷芳基、环烷基、杂环烷基、杂芳基和烷基杂芳基。更优选地,y2是芳基或杂芳基,更优选地,y2包含任选经取代的苯环,甚至更优选y2为
其中r6、r7、r8和r9彼此独立地为h或烷基,最优选h。
因此,本发明还涉及如上和如下所述的化合物和包含所述化合物的配合物,所述化合物具有以下结构:
其中a具有结构(ia)或结构(ib)。更优选地,在这种情况下,y3是o。
作为本上下文中的优选基团a,提到了以下基团:
更优选地,a选自
并且最优选地a为
根据本实施方案的优选的化合物的结构选自ca001、ca002*、ca003*、ca005*、ca006*、ca007、ca008*、ca009*、ca023*、ca025*、ca026*、ca027、ca028*和ca029*(这些化合物的结构描述于表1a)。
更优选地,该化合物的结构选自ca007、ca008*和ca009*(这些化合物的结构描述于表1a)。应当理解,如果未指定表1a中相应所示结构中的立构中心,那么这意味着所有相应立体异构体都应以单独形式以及相应立体异构体的混合物形式包括在内。因此,化合物ca002*、ca003*、ca005*、ca006*、ca008*、ca009*、ca023*、ca025*、ca026*、ca027、ca028*和ca029*可以作为单一立体异构体或立体异构体的混合物存在。更优选地,根据本实施方案,化合物的结构选自ca001、ca002、ca003、ca005、ca006、ca007、ca008、ca009、ca023、ca025、ca026、ca027、ca028和ca029(这些化合物的结构描述于表1b),其中特别优选ca007、ca008和ca009。
已经出人意料地表明,这些化合物显示出对psma的高结合亲和力并且被有效地内化。
实施方案(c)
根据本发明另一个优选的实施方案,a不是
优选地,根据本实施方案,a选自:
出人意料地发现,包含这些螯合剂构建单元的本发明的化合物与铅和/或铜的放射性核素形成稳定的配合物,并具有有利的肿瘤靶向性质。新的化合物提供了根据所用的相应放射性核素对药代动力学特征进行微调的可能性。并且,这些化合物允许用特定放射性核素进行稳定标记。
铜结合化合物:
在将化合物用作铜结合psma配体的情况下,如上所述,a优选选自
出人意料地发现,用这些化合物可以与铜放射性核素形成稳定和有效的配合物。因此,本发明还涉及如上或如下所述的化合物,或如上或如下所述的配合物,其中a是
其中放射性核素是铜放射性核素,更优选64cu和/或67cu。
例如,应该提及以下铜结合化合物,ca001、ca002*、ca003*、ca005*、ca006*、ca022*、ca023*、ca024*、ca025*和ca026*(这些化合物的结构描述于表1a)。更优选的实例是ca001、ca002、ca003、ca005、ca006、ca022、ca023、ca024、ca025和ca026(见表1b)。应当理解,如果未指定表1a中相应所示结构中的立构中心,那么这意味着所有相应立体异构体都应以单独形式以及相应立体异构体的混合物形式包括在内。因此,化合物ca002*、ca003*、ca005*、ca006*、ca022*、ca023*、ca024*、ca025*和ca026*可以以单一立体异构体或立体异构体的混合物存在。
更优选地,在将化合物与cu一起用作放射性核素的情况下,该化合物选自ca003*、ca006*、ca022*和ca023*(相应的化学结构描述于表1a),优选ca003、ca006、ca022和ca023(见表1b)。
更优选地,化合物是ca003*(见表1a),最优选为ca003(见表1b)。
出人意料地发现,利用这些化合物,可以满足psma靶向的两个尚未满足的需要:a)利用有利的生物分布特性——特别是显著改善的肾清除率实现肿瘤中的高度特异性富集;和b)能够使用铜和铅两种金属的同位素,其中存在优选的放射性同位素。
铅结合化合物:
在将化合物与例如铅一起用作放射性核素的情况下,如上所述,a优选选自
出人意料地发现,利用这种组合物,可以形成具有铅的有利的配合物,其显示有利的psma靶向性质。
因此,本发明还涉及如上或如下所述的化合物,或如上或如下所述的配合物,其中a是
其中放射性核素是铅的放射性核素,更优选203pb或212pb。
例如,应提到以下的铅结合化合物,ca007、ca008、ca009、ca010、ca011和ca012(这些化合物的结构描述于表1a),优选ca007、ca008*、ca009*、ca010*、ca011*和ca012*。
更优选地,该化合物是ca009*或ca012*,更优选ca009或ca012。因此,本发明涉及化合物或配合物,如上所述,其中该化合物的结构为ca009*或ca012(见表a1),更优选为ca009或ca012(见表1b),并且其中放射性核素优选为铅的放射性核素,更优选203pb或212pb。
出人意料地发现,这些化合物在人的血清中表现出48小时的高稳定性。此外,该化合物显示出抑制psma的高亲和力。
而且,203pb标记的化合物在psma阳性细胞系中显示了高的特异性内化率。
药物组合物
如上所述,本发明还涉及药物组合物,其包含如上或如下所述的化合物,或如上或如下所述的配合物。应当理解,药物组合物分别包含治疗有效量的化合物和/或配合物。组合物可能还包含至少一种有机或无机固体或液体和/或至少一种药学上可接受的载体。
本文使用的短语“药学上可接受的”是指在合理的医学判断范围内适合于与患者的组织接触而没有过度毒性、刺激、过敏反应或其他问题或并发症,并具有合理的获益/风险比的那些化合物、材料、组合物和/或剂型。
“患者”包括动物,例如人、猴子、牛、马、猫或狗。动物可以是哺乳动物,例如非灵长类动物和灵长类动物(例如,猴和人)。在一个实施方案中,患者为人类。
通常,式(1)的化合物或其药物组合物可以口服给药或通过肠胃外途径,通常是注射或输注给药。
“肠胃外给药途径”表示除了肠内和局部给药之外的给药模式,通常通过注射,并且包括但不限于静脉内、肌内、动脉内、鞘内、囊内、眶内、心内、皮内、腹膜内、经气管、皮下、表皮下、关节内、被膜下、蛛网膜下、椎管内和胸骨内注射和输注。
根据本发明的化合物的剂量(指载体分子的量)通过医生基于患者具体参数,例如年龄、体重、性别、疾病的严重程度等来确定。该剂量取决于施用模式:通常,用于分子成像目的的化合物以示踪剂的量来施用,即通过使用每名患者1纳摩尔至100纳摩尔的总剂量,优选的剂量为每名患者5纳摩尔至20纳摩尔。对于治疗应用(内放射疗法),需要更高剂量来实现达到治疗效果所需的辐射量吸收剂量(灰色)数。对于治疗应用,剂量优选为0.1毫摩尔/千克体重至10毫摩尔/千克体重,优选0.2毫摩尔/千克体重至5毫摩尔/千克体重,最优选0.5毫摩尔/千克体重至2毫摩尔/千克体重。对应于给药类型,药物被合适地配制,例如以溶液或混悬剂、简单的片剂或糖衣丸、硬或软明胶胶囊、栓剂、胚珠、注射用制剂的形式,其是根据常见的盖伦方法制备的。
根据本发明的化合物可以在合适的情况下与其它活性物质和药物组合物中常见的赋形剂以及载体一起配制,例如,根据要制备的制剂-滑石粉、阿拉伯树胶、乳糖、淀粉、硬脂酸镁、可可脂、水性和非水性载体、动物或植物来源的脂肪体、石蜡衍生物、二醇(特别是聚乙二醇)、各种增塑剂、分散剂或乳化剂、药学相容性气体(例如空气、氧气、二氧化碳等)、防腐剂。
为了制备液体制剂,可以使用添加剂例如氯化钠溶液、乙醇、山梨醇、甘油、橄榄油、杏仁油、丙二醇或乙二醇。
当使用用于输注或注射的溶液时,它们优选为水溶液或悬浮液,有可能在使用前制备它们,例如由含有活性物质本身或活性物质与载体如甘露醇、乳糖、葡萄糖、白蛋白等一起的冻干制剂来制备。将现成溶液灭菌,并在合适的情况下与赋形剂混合,例如与防腐剂、稳定剂、乳化剂、增溶剂、缓冲剂和/或调节渗透压的盐混合。灭菌可以通过使用具有小孔径的过滤器进行无菌过滤来实现,这种情况下,组合物可以在合适的情况下冻干。还可以添加少量的抗生素以确保无菌状态的维持。
本文使用的短语“有效量”或“治疗有效量”是指化合物、材料或包含本发明的化合物的组合物或其它活性成分的用量,所述用量能以适用于任何医学治疗的合理受益/风险比在患者的至少一个细胞亚群中有效地产生一些期望的治疗效果。关于本发明的组合物的治疗有效量是指单独的治疗剂的量,或与在治疗或预防疾病中提供了治疗益处的其它疗法组合的量。与本发明的化合物结合使用时,该术语可以包括改善整体治疗、减少或避免疾病的症状或病因、或增强其他治疗剂的治疗功效或与其他治疗剂的协同作用的量。
进一步地,本发明还涉及如上或如下所述的化合物,或如上或如下所述的配合物,或所述药物组合物,其用于治疗、改善或预防细胞增殖性疾病或病症,特别是前列腺癌和/或其转移瘤。
进一步地,本发明涉及如上或如下所述的化合物,或如上或如下所述的配合物,或药物组合物,其用于诊断。
另外,本发明涉及如上或如下所述的,或如上或下文所述的复合物,或药物组合物,其用于诊断癌症,特别是前列腺癌和/或其转移瘤。
如本文所使用的,术语“治疗”旨在还包括诊断、预防、防止、治疗和治愈。
术语“防止”是指防止由于施用预防剂或治疗剂而在患者中引起的疾病的发作、复发或传播。
优选地,将如上或如下所述的,或如上或如下所述的配合物,或药物组合物用于体内成像和放射治疗。合适的药物组合物可以含有放射显像剂,或放射治疗剂,其具有作为元素的放射性核素,即放射性碘,或足以用于成像的量的式(1a)和/或(1b)化合物的放射性金属螯合复合物,以及药学上可接受的放射性载体。放射性载体应该适合注射或抽吸,例如人血清白蛋白;水性缓冲溶液,例如三(羟甲基)-氨基甲烷(及其盐)、磷酸盐、柠檬酸盐、碳酸氢盐等;无菌水生理盐水;以及含有氯化物和/或碳酸氢盐或正常血浆阳离子如钙、钾、钠和镁的平衡离子溶液。
放射性载体中显像剂或治疗剂的浓度应足以提供令人满意的图像。例如,当使用水溶液时,剂量为0.1毫居里至300毫居里,这一宽范围是由发射α的同位素具有非常强的细胞毒性作用这一事实引起,因此在锕标记的psma-617的情况下,以低剂量例如每个治疗周期0.135毫居里225ac来应用。对于发射β的放射性同位素如177lu,通常在一个治疗周期内施用最高为216毫居里的剂量。这些剂量由技术人员确定。然而,施用至患者用于成像或治疗目的的实际剂量由实施治疗的医生决定。应施用显像剂或治疗剂以在患者中维持约1小时至10天,即使更长或更短的时间段也是可接受的。因此,可以制备含有1ml至10ml水溶液的方便安瓿。
成像可以以正常方式进行,例如通过注射足够量的成像组合物以提供充分的成像,然后用合适的成像机器或扫描机器如断层扫描仪或伽马相机进行扫描。在某些实施方案中,对患者体内的区域进行成像的方法包括以下步骤:(i)将诊断有效量的与放射性核素络合的化合物施用至患者;将患者的区域暴露于扫描装置并(ii)获得患者该区域的图像。在某些实施方案中,成像的区域是头或胸。在其他实施方案中,式l(a)和/或式(lb)的化合物或配合物靶向psma蛋白。
因此,在一些实施方案中,提供了使组织如脾组织、肾组织或表达psma的肿瘤组织成像的方法,所述方法包括使组织与配合物接触,所述配合物通过将放射性核素和式(la)和/或式(lb)的化合物接触来合成。
施用至患者的本发明的化合物或含有化合物的配合物的制剂或其盐、溶剂化物、立体异构体、或互变异构体的量取决于几种生理因素。这些因素是医生已知的,包括要执行的成像的性质、要用于成像或治疗的目标组织以及要使用放射性药物成像或治疗的患者的体重和病史。
根据另一个方面,本发明提供了一种治疗患者的方法,通过给予患者治疗有效量的如上所述的复合物来治疗患有细胞增殖性疾病或病症的患者。特别地,使用根据本发明的化合物、药物组合物或放射性药物治疗或成像的细胞增殖性疾病或病症是癌症,例如,前列腺癌和/或在例如肺、肝脏、肾脏、骨、脑、脊髓、膀胱等中的前列腺癌转移瘤。
在实施例章节中详细描述了本发明的化合物的合成。
概括本发明发现的以下实施方案是特别优选的:
1、式(1)的化合物:
或其药学上可接受的盐或其溶剂化物,
其中y3是o或s,
其中s、t和w彼此独立地为0或1,
其中i是1到3的整数,
其中j是3到5的整数,
并且其中z1、z2和z3彼此独立地选自co2h、-so2h、-so3h、-oso3h和-opo3h2,
r1为-ch3或h,优选为h,
x选自任选经取代的烷基芳基(-烷基-芳基)、芳基、烷基杂芳基(-烷基-杂芳基)和杂芳基,
y1和y2彼此独立地选自任选经取代的芳基、烷基芳基(-烷基-芳基)、环烷基、杂环烷基、杂芳基和烷基杂芳基(-烷基-杂芳基),
并且其中a是螯合剂残基,其结构选自(ia)、(ib)和(ic)
其中r2、r3、r4和r5彼此独立地选自h、-ch2-cooh和-ch2-c(=o)-nh2或其中r2和r4形成-(ch2)n-桥,n为1至3的整数,其中n优选为2,
其中r、v和q彼此独立地为0或1,
条件是在u和w为0的情况下,q和v为0,并且
(a)其中u和w为1,或
(b)其中u为0和w为1,并且其中a选自(ia)或(ib),或
(c)a不是
2、根据实施方案1所述的化合物,其中x优选包含选自任选经取代的萘基、苯基、联苯基、吲哚基和苯并噻唑基的残基,更优选地,其中x选自
3、根据实施方案1所述的化合物,其中x是
4、根据实施方案1至3中任一项所述的化合物,其中z1、z2和z3是-co2h。
5、根据实施方案1至4中任一项所述的化合物,其中r1是h。
6、根据实施方案1至5中任一项所述的化合物,其中y1是
7、根据实施方案1至6中任一项所述的化合物,其中i为2且j为4,其中化合物优选具有结构(1a)
8、根据实施方案1至7中任一项所述的化合物,其中u和w为1。
9、根据实施方案8所述的化合物,其中y3是s。
10、根据实施方案8或9所述的化合物,其中y2是
其中r6、r7、r8和r9彼此独立地为h或烷基,优选h。
11、根据实施方案8至10中任一项所述的化合物,其中r优选为0。
12、根据实施方案8至11中任一项所述的化合物,其中a是螯合剂,其选自:
13、根据实施方案8至12中任一项所述的化合物,其结构选自ca007、ca008*、ca009*、ca029*和ca030*(见表1a),优选结构选自ca007、ca008、ca009、ca029和ca030(见表1b)。
14、根据实施方案8至12中任一项所述的化合物,其结构选自ca007、ca008*和ca009*(见表1a),优选结构选自ca007、ca008和ca009(见表1b)。
15、根据实施方案1至7中任一项所述的化合物,其中u为0且w为1,其中a选自(ia)或(ib)。
16、根据实施方案15所述的化合物,其中y2是任选经取代的芳基或杂芳基。
17、根据实施方案16或17所述的化合物,其中y2是
其中r6、r7、r8和r9彼此独立地为h或烷基,优选h。
18、根据实施方案15至17中任一项所述的化合物,其中a选自:
19、根据实施方案15至18中任一项所述的化合物,其中a选自
20、根据实施方案15至19中任一项所述的化合物,其中a是
21、根据实施方案15至20中任一项所述的化合物,其结构选自ca001、ca002*、ca003*、ca007、ca008*、ca009*、ca022*、ca023*、ca025*、ca026*、ca027、ca028*和ca029*(见表1a),优选结构选自ca001、ca002、ca003、ca007、ca008、ca009、ca022、ca023、ca025、ca026、ca027、ca028和ca029(见表1b)。
22、根据实施方案15至21中任一项所述的化合物,其结构选自ca007、ca008*和ca009*,优选结构选自ca007、ca008和ca009(表1b)。
23、根据实施方案1至7中任一项所述的化合物,其中a不是
24、根据实施方案23所述的化合物,其中a选自
25、配合物包括
(a)放射性核素,和
(b)根据权利要求1至24中任一项所述的化合物或其盐。
26、根据实施方案25所述的配合物,其中放射性核素选自89zr、44sc、111in、90y、66ga、67ga、68ga、177lu、99mtc、60cu、61cu、62cu、64cu、66cu、67cu、149tb、152tb、155tb、161tb、153sm、153gd、155gd、157gd、213bi、225ac、230u、223ra、165er、fe的放射性核素(例如52fe和59fe)和pb的放射性核素(例如203pb和212pb、211pb、213pb、214pb、209pb、198pb、197pb)。
27、根据实施方案25所述的配合物,其中放射性核素是铅的放射性核素,并且a选自
28、根据实施方案27所述的配合物,其中(b)是结构选自ca007、ca008、ca009、ca010和ca011或其盐的化合物。
29、根据实施方案25所述的配合物,其中放射性核素是铜的放射性核素,并且a选自
30、一种药物组合物,其包含实施方案1至24中任一项所述的化合物或权利要求书25至29中任一项所述的配合物。
31、实施方案1至24中任一项所述的化合物或实施方案25至29中任一项所述的配合物或实施方案30所述的药物组合物,其用于治疗、改善或预防表达psma的癌症和/或其转移瘤,特别是前列腺癌和/或其转移瘤。
32、实施方案1至24中任一项所述的化合物或实施方案25至29中任一项所述的配合物或实施方案30的药物组合物,其用于在诊断。
33、实施方案1至24中任一项所述的化合物或实施方案25至29中任一项所述的配合物或实施方案30所述的药物组合物,其用于诊断癌症,例如表达psma的癌症和/或其转移瘤,特别是前列腺癌和/或其转移瘤的用途。
本说明书全文引用的所有参考文献具体提及的公开内容及其全部内容通过引用结合于此。
附图
图1:表1a:优选的化合物的概述。应当理解,如果未指定相应所示结构的立构中心,那么这意味着所有相应立体异构体都应以单独形式以及相应立体异构体的混合物形式包括在内,表1b:非常优选的化合物的概述。
图2:合成psma-螯合剂缀合物的反应方案(a)三光气、dipea、ch2cl2,0℃;(b)h-lys(alloc)-2ct-树脂,ch2cl2;(c)pd[p(c6h5)3]4,吗啉,ch2cl2(d)fmoc-2-nal-oh,hbtu,dipea,dmf;(e)20%哌啶,dmf;(f)反式-4-(fmoc-氨甲基)-环己烷甲酸,hbtu,dipea,dmf;(g)20%哌啶,dmf;(h)螯合剂,hbtu(如果需要的话),dipea,dmf;(i)95%的tfa,2.5%h2o,2.5%tips。
图3:表中所示的核素标记的新的psma配体的psma抑制能力和特异性内化值。使用psma阳性的c4-2细胞系进行竞争性细胞结合。与psma-617相比,新的配体在低纳摩尔范围内的ki显示出良好抑制作用,并且部分具有更高的内化值(特异性裂解物)。
图4:a.显示64cu-psma-617、64cu-ca003和64cu-ca023生物分布的c4-2荷瘤小鼠的pet扫描和时间-活性曲线。64cu-psma-617在肿瘤和肾脏以及肝脏中显示出很高累积。a.64cu-psma-617、64cu-ca003和64cu-ca023在不同时间点(0分钟至20分钟、40分钟至60分钟、2h、48h)的生物分布可视化。b.显示64cu-psma-617和64cu-ca003生物分布的c4-2荷瘤小鼠的动态pet扫描(0分钟至60分钟)时间-活性曲线。肿瘤和肝脏的标准摄取值(suv)显示,肝脏中64cu-psma-617的摄取量高于肿瘤,与64cu-ca003相反。
图5:在不同时间点(0h至48h)c4-2荷瘤小鼠的pet图像中64cu-psma-617、64cu-ca003和64cu-ca023的最大标准摄取值(msuv)。64cu-ca003显示了在肿瘤和肾脏中的高积累,64cu-ca023显示了在肿瘤中的高积累以及从肾脏中的快速清除。
图6a:在c4-2荷瘤小鼠解剖的目的器官中累积64cu-ca003证实了肿瘤特异性和从肾脏中的快速清除。
图6b:在c4-2荷瘤小鼠解剖的目的器官中累积64cu-ca023证实了肿瘤特异性和从肾脏的快速清除。
图6c:在以下时间点,0.025纳摩尔的64cu-ca003的器官分布:注射后10分钟、1h、4h、24h和72h。值表示为%id/g组织±标准差;所有组织的n=3。
图6d:在阻断实验(b)中,放射性示踪剂64cu-ca003(0.030纳摩尔)与每千克体重2mgpsma-617同时注射。值表示为%id/g组织±标准差;所有组织的n=3。
图7:患者注射后2小时(a)和20小时(b)的64cu-ca003(200mbq,0.5纳摩尔)pet/ct最大强度投影。红色箭头指向选定的来自肩胛骨来源的右肩软组织浸润,随着时间变化,肺、骨骼和淋巴结转移瘤的对比增加。肝胆清除导致肠内出现热点;必须提供横截面切片(c),以避免出现假阳性读数。
图8:在balb/cnu/nuc4-2荷瘤小鼠中尾静脉注射后1h(a)不同203pb标记的化合物的平面闪烁照相成像以及(b)203pb-ca012的分布时间进程。ca009和ca012的放射性标记衍生物显示示踪剂在肿瘤组织中的高摄取。测定的203pb-ca012摄取动力学显示放射性示踪剂在肿瘤组织中的保留时间较长。当与203pb-ca009相比时,203pb-ca012摄取的选择性增强。这是非靶器官快速清除的结果。
图9:203pb-psma-ca012在荷瘤小鼠中的器官分布,0.025纳摩尔的203pb-psma-ca012。这种定量确认了成像实验的结果。肿瘤与肾脏摄取的高比率是由于肾脏的排泄值较高引起的。
图10:203pb-ca012平面扫描随时间变化的几何平均值图像(a)与利用177lu-psma-617的治疗扫描(b)对比;两者均使用介质能量准直器取得。
图11:基于olinda中雄性-成年模型的诊断203pb-ca012(左列)和治疗212pb-ca012(右列)的安全剂量测定估计值(uli=上大肠,lli=下大肠)。
图12:对唾液腺、随机选择的肿瘤病变(球形模型)以及假定的剂量限值器官的212pb-ca012(“tcmc”-psma-617)剂量测定与213bi-psma-617和225ac-psma-617的对比。
图13a:注射后1h和3h多发性淋巴结前列腺癌转移瘤的68ga-psma-ca028pet扫描的最大强度投影。横断面切片显示腋下和肺门(c)淋巴结转移瘤(由红色箭头指示);也可在作为参考标准的相关ct上描绘。
图13b:注射295mbq/20纳摩尔68ga-ca030后1h(a)和3h(b)进行的psma-pet最大强度投影。箭头指向显示轴向骨架(c至e)的多个区域中骨转移瘤的横截面切片位置。在ct(f)中,仅通过形态信息,没有典型的成骨细胞反应可以描绘肿瘤轮廓。
图14:以注射后1h、2h和4h68ga-psma-ca028的以%id/g组织±sd(n=3)表示的器官分布。
图15:在带有c4-2肿瘤异种移植物的balb/cnu/nu小鼠中,所选68ga-psma配体在注射后2h的全身小动物pet成像(a)与68ga-ca028的时间进程(b)和68ga-ca030的时间进程(c)的比较。
图16:通过放射-itlc法测定177lu-ca028、177lu-ca029、177lu-ca030与177lu-psma-617在37℃下72h的血清稳定性(平均±sd,n=3)。
图17:通过放射-itlc法测定64cu-ca003、64cu-ca005和64cu-psma-617在37℃下72h的血清稳定性(平均±sd,n=4)。
图18:通过活性测量测定64cu-ca003、64cu-ca005和64cu-psma-617在37℃下72h的血清稳定性(平均±sd,n=4)。
图19:64cu-ca003在balb/c裸鼠(无肿瘤)中10分钟p.i.的体内代谢物分析。来自肾脏、血液和肝脏的提取物的放射-hplc色谱图显示,活性在完整示踪剂的保留时间洗脱。这证明了铜配合物在主要分布期内的完整性。
图20:balb/c裸鼠(无肿瘤)中在10分钟p.i.时肝脏中64cu-ca003提取物的放射-hplc色谱图与肝脏中64cu-氯化物的比较。
图21a:带有c4-2肿瘤异种移植物的balb/cnu/nu小鼠的全身小动物pet扫描作为最大强度投影。64cu-psma-617(10mbq,0.2纳摩尔)、64cu-psma-ca003(10mbq,0.2纳摩尔)的pet成像。
图21b:64cu-psma-ca003(5mbq,0.030纳摩尔)与过量的未标记psma-617(2毫克/千克体重)和64cu-氯化物(10mbq)共同注射。颜色条给出了suv和pet图像色阶之间的关联,0=最小值,4=最大值。
图22:带有c4-2肿瘤异种移植物的balb/cnu/nu小鼠的全身小动物pet扫描作为最大强度投影的比较。用68ga(20mbq,0.2纳摩尔)放射性标记的四个新psma配体的注射后2h的pet成像(a),68ga-ca028(b)的时间进程和68ga-ca030(c)的时间进程。颜色条给出了suv和pet图像色阶之间的关联,0=最小值,4e0=最大值。
图23:68gapsma-ca027(0.6纳摩尔,5mbq)和68gapsma-ca028(0.6纳摩尔,6mbq)的血液时间-活性曲线,包括双指数曲线拟合。
图24:在balb/c裸鼠(肿瘤)中1hp.i.的177lu-ca028与177lu-psma-617(10mbq,0.2纳摩尔在约100μl的0.9%盐水中)的体内代谢物分析对比。来自肾脏、血液、肝脏和肿瘤的提取物的放射-hplc色谱图显示,活性在完整示踪剂的保留时间洗脱。这证明了配合物在主要分布期内的完整性。
图25:注射后20分钟、1h、2h和4h,0.05纳摩尔68ga-ca028的器官分布,以%id/g组织±sd(n=3)表示。
图26:用64cu标记的新化合物的放射-hplc色谱图。
图27:用68ga标记的新psma配体的时间-活性曲线。注射后1h内(a)肾脏的时间-活性曲线和(b)肿瘤的时间-活性曲线。数据为平均标准化摄取值(suv平均值)。
图28:(a)在雌性swiss小鼠中注射9mbq(0.30纳摩尔)64cu-ca00310分钟后的pet图像。最大强度投影(mip)表示血液中的循环和肾脏的摄取。(b)在雌性swiss小鼠中注射10mbq64c10分钟p.i.后的pet图像。最大强度投影(mip)表示肝脏和肾脏中的强摄取。
以下实施例仅用于说明本发明。无论怎样,它们不应被解释为限制本发明的范围。
实施例:
材料和方法
溶剂和化学品从merck(德国达姆施塔特)和sigma-aldrich(德国慕尼黑)购得,无需进一步纯化即可使用。体外实验进行三次,每次进行实验至少获得三组独立数据。前列腺癌患者的pet成像由海德堡大学医院根据德国法律批准,并获得赫尔辛基宣言的批准(许可s321/2012)。
螯合剂部分的合成
螯合剂部分以高产率合成,并通过lc-ms进行表征。螯合剂双官能大环环拉胺类似物4-[(1,4,8,11-四氮杂环十四烷胺-1-基)-甲基]苯甲酸的合成由studer和kaden进行了描述(studerm,andkadan,t.a.one-stepsynthesisofmono-n-substitutedazamacrocycleswithacarboxylicgroupintheside-chainandtheircomplexeswithcu2+andni2+.helvetica.1986;69:2081–2086),而交联桥螯合剂4-羧甲基-11-(1,3-二羧基丙基)-1,4,8,11-四氮杂双环[6.6.2]十六烷-戊二酸由boswell等人进行了报导(boswellca,reginoca,baidooke等人,synthesisofacross-bridgedcyclamderivativeforpeptideconjugationand64curadiolabeling.bioconjugchem.2008;19:1476-1484)。
i.一般步骤:新psma配体的合成
psma结合模体在2-氯三苯甲基树脂(2ct-树脂)上通过固相合成来制备,如eder等人(ederm,
ii.用于铜同位素成像和治疗的配体
(ca001)的说明
通过在500μl的dmf中将树脂(化合物4)与1.5当量的ctpa-nhs-酯(4-[(1,4,8,11-四氮杂环十四烷-1-基)-甲基]苯甲酸)和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:1.68分钟;esi-ms(m/z):[m+h]+(c30h50n7o8计算值):636.37(636.36),
螯合剂ctpa-nhs-酯的化学结构,用于合成ca001、ca002和ca003的化合物。
ca002的说明
通过在500μl的dmf中将树脂(化合物5)与1.5当量的ctpa-nhs-酯(4-[(1,4,8,11-四氮杂环十四烷-1-基)-甲基]苯甲酸)和10当量的二异丙胺(dipea)一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.39分钟;esi-ms(m/z):[m+h]+c43h61n8o9计算值):833.42(833.45)。
ca003的说明
通过在500μl的二甲基甲酰胺(dmf)中将树脂(化合物6)与1.5当量的ctpa-nhs-酯(4-[(1,4,8,11-四氮杂环十四烷-1-基)-甲基]苯甲酸)和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.50分钟;esi-ms(m/z):[m+h]+(c51h74n9o10计算值):972.52(972.55)。
ca005的说明
通过在500μl的dmf中将树脂(化合物5)与1.5当量的交联桥-te2a螯合剂、0.98×n螯合剂hbtu和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.38分钟;esi-ms(m/z):[m+h]+(c44h64n8o13计算值):913.45(913.47),
螯合剂8-羧甲基-交联桥-te2a的化学结构,用于合成ca005和ca006的化合物。
ca006的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的交联桥-te2a螯合剂、0.98×n螯合剂hbtu和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.55分钟;esi-ms(m/z):[m+h]+(c52h78n9o14计算值):1052.62(1052.56)。
ca022的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的交联桥-ctpa螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.72分钟;esi-ms(m/z):[m+h]+(c53h76n9o10计算值):998.56(998.57),
螯合剂交联桥-ctpa的化学结构,用于合成ca022的化合物。
ca023的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的8-羧甲基-ctpa螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.54分钟;esi-ms(m/z):[m+h]+(c53h76n9o12计算值):1030.55(1030.56),
螯合剂8-羧甲基-ctpa的化学结构,用于合成ca023的化合物。
ca024的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的8-羧甲基-交联桥-ctpa螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.60分钟;esi-ms(m/z):[m+h]+(c55h78n9o12计算值):1056.56(1056.57)
螯合剂8-羧甲基-交联桥-ctpa的化学结构,用于合成ca024的化合物。
ca025的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的8,11-双(羧甲基)-ctpa螯合剂[cpta=4-[(1,4,8,11-四氮杂环十四烷-1-基)甲基]苯甲酸]和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.60分钟;esi-ms(m/z):[m+h]+(c55h78n9o14计算值):1088.55(1088.56),
螯合剂8,11-双(羧甲基)-ctpa的化学结构,用于合成ca025的化合物。
ca026的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的8,11-双(羧甲基)-ctpa螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.53分钟;esi-ms(m/z):[m+h]+(c57h80n9o16计算值):1146.56(1146.57),
螯合剂4,8,11-三(羧甲基)-ctpa的化学结构,用于合成ca026的化合物。
iii.用于α疗法的铅同位素(203pb/212pb)的psma配体
ca007的说明
通过在500μl的dmf中将树脂(化合物5)与1.5当量的p-scn-bn-tcmc螯合剂[tcmc=1,4,7,10-四氮杂-1,4,7,10-四(2-氨甲酰基甲基)环十二烷]和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.41分钟;esi-ms(m/z):[m+h]+(c49h70n13o12s计算值):1064.49(1064.50),
螯合剂p-scn-bn-tcmc的化学结构,用于合成ca007、ca008和ca009的化合物。
ca009的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的p-scn-bn-tcmc螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。esi-ms(m/z):[m+h]+(c57h83n14o13s计算值):1203.59(1203.60)。
ca011的说明
通过在500μl的dmp中将树脂(化合物5)与1.5当量的2-(4,7,10-三(2-氨基-2-氧代乙基)-1,4,7,10-四氮杂环十二烷-1-基)乙酸、螯合剂1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酰胺(do3am)的单羧酸酯衍生物、0.98×n螯合剂hbtu和10当量的dipea一起培育,来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.07分钟;esi-ms(m/z):[m+h]+(c41h62n11o12计算值):900.45(900.46),
螯合剂的化学结构
2-(4,7,10-三(2-氨基-2-氧乙基)-1,4,7,10-四氮杂环十二烷-1-基)乙酸,螯合剂1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酰胺(do3am)的单羧酸酯衍生物,用于合成ca010、ca011和ca012的化合物。
ca012的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的do3am螯合剂、0.98×n螯合剂hbtu和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.21分钟;[m+h]+(c49h75n12o13计算值):1039.54(1039.56)。
iv.增强psma-617药代动力学性质的螯合剂间隔子部分
ca027的说明
通过在500μl的dmf中将树脂(化合物4)与1.5当量的p-nhs-bn-dota螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:1.47分钟;[m+h]+(c34h52n7o14计算值):782.33(782.36),
p-nhs酯-bn-dota的化学结构,用于合成ca027和ca028的螯合剂。
ca028的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的p-nhs-bn-dota螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.36分钟;[m+h]+(c55h76n9o16计算值):1119.53(1118.54)。
ca029的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的p-scn-bn-dota螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.49分钟;[m+h]+(c57h79n10o17s计算值):1207.52(1207.53),
螯合剂p-scn-bn-dota的化学结构,用于合成ca029的化合物。
ca030的说明
通过在500μl的dmf中将树脂(化合物6)与1.5当量的p-ncs-苄基-dota-ga螯合剂和10当量的dipea一起培育来获得产物。如上所述,纯化该化合物并通过hplc分析最终产物(见章节i)。hplc-保留时间:2.50分钟;[m+h]+(c60h84n11o18s计算值):1278.56(1278.57),
螯合剂p-ncs-苄基-dota-ga的化学结构,用于合成ca030的化合物。
v放射性标记配合物的合成
v.164cu-psma-衍生物的放射化学合成
将缀合物(水中的1mm,5μl,5纳摩尔)添加至400μl醋酸钠缓冲液(水中的0.4m,ph5.0)、10μl抗坏血酸(20%的水溶液)和282μl[64cu]cucl2(于0.1m的hcl(200mbq))的混合物中。在95℃下加热混合物5分钟。通过放射-hplc(0%至100%mecn进行5分钟,monolith色谱柱)控制标记,流速为2毫升/分钟,保留时间为2.3分钟。
该标记导致10分钟内放射标记产率>98%(如图26中放射色谱图所示)。例如,64cu-psma-ca003的比放射性约为40兆贝可/纳摩尔。将相同的方案用于67cu的标记。
v.2203/212pb-psma-配体的放射化学合成
80纳摩尔缀合物(水中的1mm,80μl,80纳摩尔)添加至400μl醋酸钠缓冲液(水中的0.4m,ph5.0)、10μl抗坏血酸(水中的20%)和140μl203pb-氯化物溶液(0.04mhcl)中,其中比放射性约为102.6tbq。(lantheusmedicalimaging,美国)。然后,在95℃下将混合物加热5分钟。通过放射-hplc控制标记。
v.368ga-psma-ca028(ca027、ca029、ca030)的放射化学合成
从68ge/ga生成器(ithembalabs,南非)中洗脱68ga。将缀合物(1mm在dmso,20μl,20纳摩尔)添加到320μl醋酸钠缓冲液(水中的0.4m,ph4至5)、10μl抗坏血酸(20%的水溶液)和400mbq68ga(0.6m的hcl)中。在95℃下加热混合物5分钟。通过放射-hplc(0%至100%mecn进行5分钟,monolith色谱柱)控制标记,其中流速为2毫升/分钟,保留时间为2.4分钟。
选定的新配体的分析数据
v.4177lu-psma-ca028(ca027、ca029、ca030)的放射化学合成
对于177lu标记,将约20mbq与200μl含有chelex的0.4m醋酸钠缓冲液(ph=5)混合。将2μl在10%dmso的水溶液中的1mm化合物溶液、2μl抗坏血酸饱和溶液和40μl[177lu]lucl3溶液进行混合,并加热至95℃并保持10分钟。通过放射-hplc(水中的0%至100%acn进行5分钟,monolith色谱柱)检查标记。
vi.临床前评估
使用psma阳性c4-2细胞系,即lncap(前列腺淋巴结癌)细胞系的亚系(crl-3314;americantypeculturecollection)来进行体外和体内实验。在补充有10%胎牛血清和稳定谷氨酰胺(panbiotech)的rpmi1640(panbiotech)培养基中培养c4-2细胞。细胞在37℃下生长并用与5%co2平衡的湿润空气孵育。
vi.1体外
vi.1.1竞争性结合试验和内化率
在室温下,使用每个孔含1%bsa的100μlpbs对multiscreenhts-dv滤板培养30分钟。去除pbs/bsa溶液之后,向opti-mem的每个孔添加1×105个c4-2细胞。使用0.75nm的68ga标记的psma-hbed-cc二聚体(68ga-psma-10)(
对于测定特定的内化率,在室温下用在pbs中0.1%的多聚-l-赖氨酸对24孔板孵育20分钟,并使用pbs清洗一次。在下一步,向每个孔添加含有1×105个c4-2细胞的1mlrpmi介质并培养过夜。实验期间每种化合物的条件是:在37℃或4℃下通过终浓度为500μm的2-(膦酰基甲基)戊二酸(2-pmpa;axxora)阻断或不阻断受体的条件下孵育。然后,用250μl的30nm标记化合物溶液孵育细胞。将板在37℃的水浴中或在4℃的冰上孵育45分钟。然后,用1ml冰冷的pbs将细胞清洗3次,并用甘氨酸(于hcl中的50mm,ph2.8)孵育5分钟。利用1ml冰冷的pbs进行额外的清洗步骤之后,用0.5ml的0.3mnaoh使细胞溶解,收集并用γ计数器测量放射性1分钟。通过减去阻断条件下的相应摄取量,以结合至106个细胞(%ia/106个细胞)的初始添加放射性的百分比测定特定细胞摄取。所有实验进行三次。
结果示于图3。ki测定显示合成配体对psma的纳摩尔结合亲和力。
对于203pb标记的化合物,化合物ca009和ca012显示出抑制psma的最高亲和力。而且,203pb标记的化合物ca009和ca012在psma阳性细胞系中显示出较高的特异性内化率。化合物203pb-ca009和203pb-ca012显示内化率最高为28.36±2.23,注射活性为7.33±1.26/106个c4-2细胞(n=3)。
如图3所示,对于cu标记的配体,例如ca003,显示出对psma的特别高的亲和力,然后是ca006、ca002和ca026。而且,64cu标记的化合物显示出对c4-2细胞的特异性结合。64cu-ca003内化了34.63±2.77%,64cu-ca005内化了18.63±4.46%,64cu-ca022内化了、38.7±6.69%(n=3)。106个c4-2细胞被用于这些实验。
进一步地,例如ga标记的配体的ki测定结果显示例如合成配体对psma的纳摩尔结合亲和力。如图3所示,在所有新化合物中,ca030显示出特别有利的性能。
vi.1.2血清稳定性
vi.1.2.1203pb标记的化合物的血清稳定性
通过在37℃的300μl人血清中孵育1h、2h、3h、6h、24h、48h和72小时后来测定放射性标记的化合物的稳定性。通过加入2份乙腈沉淀血清。然后,涡旋样品并在13000rpm下离心5分钟(2次),通过放射-hplc(0%至100%mecn进行5分钟,monolith色谱柱)分析上清液。
所有化合物在人血清中显示出至少48小时的高稳定性。ca011和ca012显示出特别有利的稳定性,并且稳定至少72小时。
vi.1.2.268ga、177lu和64cu标记的化合物的血清稳定性
在化合物的放射性标记后,通过itlc和hplc分析测定血清稳定性。向200μl人血清(h4522;sigma-aldrich,德国)中添加50μl(20mbq)标记的配体,并在37℃下培养不同的时间点(0h、2h、24h、48h和72h)。使用0.5×5cm的itlc-sg-玻璃微纤维色层分析纸(folsom,美国加州)。将0.5μl血清中的放射性标记的化合物施加到距底部(原点)1cm处的每个条带,并使溶剂(在177lu配体的情况下为柠檬酸钠缓冲液(0.5m,ph=5.0),在cu标记的配体的情况下为(1%na-edta,ph=4))前沿从底部上升到5cm。最后,将每个纸条分成8份;每份在γ计数器中测量。对于hplc分析,向样品中添加等体积的acn以沉淀血清蛋白。然后,使样品以130000rpm离心10分钟,分离沉淀和上清液,并且测量相对活性。结果以百分比表示。另外,通过放射-hplc(0%至100%acn进行5分钟,monolith色谱柱)分析上清液的等分样品,其中流速为2毫升/分钟。
用68ga和177lu放射性标记的化合物的稳定性测试结果示于图16。如hplc和放射tlc所示,68ga标记的化合物在人血清中孵育2h后未显示降解。在孵育24小时后177lu-ca028显示40%的游离177lu,而在这个时间点ca029、ca030和参考化合物psma-617未显示游离活性(图16)。
64cu标记的化合物的稳定性测试结果示于图17和图18。在孵育2h后,itlc显示所有化合物仅解离低于2±0.6%。在孵育24h后,发现仅6±4%的64cu-ca003和3±1%的64cu-ca006解离。相反,64cu-psma-617显示了13±3%的游离64cu活性。长期稳定性测试(72h)显示64cu-ca006(8±4%)具有与64cu-ca003(11±3%)相当的稳定性。在这个时间点,发现18±6%的64cu-psma-617解离。在孵育2h后,沉淀中的活性测量(图18)显示19.0±5.2%的64cu-ca003、12±7.2%的64cu-ca006以及40±7.5%的64cu-psma-617与蛋白组分一起沉淀。沉淀中活性的百分比随时间增加。在64cu-psma-617的沉淀中测量到最高活性量,然后是64cu-ca003,并且发现64cu-ca006沉淀活性较低。
vi.2体内实验
根据德意志联邦共和国法律进行体内实验。对于pet成像和生物分布研究,从charlesriver获得4周至5周大的雄性裸鼠(balb/cnu/nu小鼠)(19g至23g),并在研究前在无特定病原体条件下保存1周。这些老鼠被安置在12小时/12小时的光照/黑暗周期中,并且可以自由地获得水和食物。小鼠用2%七氟醚麻醉,然后在右侧躯干皮下接种培养在opti-memi(1×)培养基中的50%基质胶中的5×107个c4-2细胞。当肿瘤尺寸约为1cm3时,进行器官分布研究。
vi.2.1铅标记的化合物:
vi.2.1a闪烁成像和生物分布
对于小动物成像,小鼠用2%七氟醚麻醉。向尾静脉注射0.1纳摩尔(1.0mbq)的203pb-配体。在10分钟、1h、4h、24h和72h后,使用带有平行准直仪(35mm/1.8mm/0.2mm)的γ成像仪sct(biospacelab,法国巴黎)进行连续平面扫描。基于成像结果,选择化合物ca012进行生物分布研究。实验进行三次。
结果在图8和图9中给出。
vi.2.264cu标记的化合物:
vi.2.2a)64cu-氯化物和64cu-ca003的在血液中的稳定性和体内命运
通过itlc和hplc测定64cu标记的ca003的体内稳定性。无瘤雄性balb/c裸鼠(n=3)经尾静脉注射64cu-ca003(3.6mbq;0.26纳摩尔,溶解在总体积约为100μl的0.9%盐水中)并且在注射后10分钟收集800μl血液。将血液样本以13000rpm离心10分钟。然后,分离沉淀与上清液,并测定相对活性。进行itlc以评估放射性标记的化合物在如上所述的血液中的稳定性。而且,在加入等体积的acn并通过离心去除蛋白质后,通过放射-hplc(0%至100%acn进行5分钟,monolith色谱柱)分析上清液等分样品,其中流速为2毫升/分钟。
通过放射-hplc分析研究体内新陈代谢。无瘤雌性swiss小鼠(n=3)经尾静脉注射64cu-氯化物(10mbq,在约100μl的0.9%盐水中)或64cu-ca003(9mbq,0.30纳摩尔,在约100μl的0.9%盐水中)。在注射10分钟后进行pet成像,然后收集血液、肝脏和肾脏。用预冷却的盐水冲洗组织,吸干并用2ml的0.1mnh4oac/etoh(35∶65)处理。使用ultra-turraxt8(ikalabortechnik,德国)对组织进行匀浆。使样本以13000rpm(4℃)离心10分钟。然后,分离沉淀与上清液,并测量相对活性。结果以百分比表示。另外,如上所述,通过用acn沉淀蛋白质,制备上清液等分样品用于hplc测量。另外,通过放射-hplc(0%至100%acn进行5分钟,monolith色谱柱)分析样品,其中流速为2毫升/分钟。在整个色谱分析过程中,每10秒采集组分,并在γ计数器中测量样本的相对活性,以重建色谱图。
血液稳定性的itlc结果显示,64cu-ca003经历3%的64cu解离或97±2.3%的完整示踪剂(见图19)。放射hplc色谱图同样证实了铜配合物的完整性(见图20)。进行64cu-氯化物或64cu-ca003体内命运研究,64cu-氯化物和64cu-ca003的pet成像显示出不同的药代动力学(图s21)。64cu-氯化物的最大强度投影pet成像显示了较低的血液循环(1.3)、高的肝脏摄取(2.7)和肾脏(3.4)摄取。而64cu-ca003显示出比64cu-氯化物更长的血液循环(2.3)、更高的肾脏(5.7)摄取和更低的肝脏积累(1.0)(图28)。肾脏、血液和肝脏组织提取物的放射hplc色谱图证明了64cu-ca003的完整性(图19)。64cu-ca003的色谱图显示示踪剂的保留时间不同于游离铜64cu-氯化物(图20)。
vi.2.2b)器官分布实验(64cu配体)和小动物pet
基于pet成像结果,选择ca003和ca023使用c4-2荷瘤小鼠进行生物分布分析。实验进行三次。通过尾静脉注射给予0.025纳摩尔64cu标记的化合物(在约100μl的0.9%盐水中,每只小鼠1mbq)。在时间点:10分钟、1h、4h、24h和72h,解剖器官并称重,使用γ计数器(packardcobraauto-gamma)测量活性。计算每克注射剂量的百分比(%id/g)。(见图6a、图6b)
除此之外,显示了在1h(n=3)同时施用psma-617以阻断psma结合的实验(图6c和图6d)。
为进行利用各种64cu标记的psma配体的小动物pet成像,0.2纳摩尔、10mbq的约100μl0.9%盐水中的放射性标记化合物注射到c4-2荷瘤小鼠中。在小动物pet扫描仪(siemensinveond-pet,malvern,美国宾夕法尼亚州)中记录动态pet。从常规(非动态)pet图像中获得suv值。suv的公式为:
目的体积(voi)是通过手动描绘适当的整个组织(心脏、肾脏、膀胱、肿瘤,体积约为100μl至500μl)或部分组织肝脏和肌肉获得的。根据以下程序重建图像:osem3d/spmap,含16个子集,2次迭代,图像x-y大小:256,图像z大小:161。未使用后处理过滤器修改数据。用于分析图像和tac的软件是来自siemensirw4.1的inveontmacquisitionworkplace(iaw)。注射后进行动态pet扫描0分钟至60分钟,在20分钟(0分钟至20分钟、20分钟至40分钟和40分钟至60分钟)的三个时间帧中重建图像用于视觉显示。如图2/图3和表s1+s2所示,对于一些显示出长保留时间的化合物,包括后来的时间点(2h、4h、20h、45/48h)。1h后,进行静态pet扫描。为了比较不同放射性示踪剂,描绘了随时间的平均suv。
图6a至6d显示了psma配体64cu-ca003(n=3)获得的生物分布结果。注射后10分钟,观察到11.33±4.11%id/g肿瘤的摄取。4h后,肿瘤中示踪剂积累的量(32.34±10.6%id/g)远高于肾脏中积累的量(13.33±3.36%id/g)。从动态pet成像中产生的时间活性曲线显示,在注射一小时后,肿瘤与肌肉的比为10.5,肿瘤与血液的比为3.0,见下表vi.2.2b_1:
表vi.2.2b_1在携带c4-2肿瘤异种移植物的balb/cnu/nu小鼠中,从64cu-ca003小动物pet获得的时间-活性曲线得出的平均标准化摄取值(msuv)。
这些曲线表明肾脏的迅速摄取。器官分布研究显示,在1h时高的肾脏摄取量(67.04±20.89%id/g)在24h内大部分清除(7.48±8.51%id/g)。相反,高肿瘤摄取值(30.83±12.61%id/g于1hp.i.)几乎保持不变(19.99±6.43%id/g于24hp.i.)。pet成像证实了放射性示踪剂在肿瘤中的强积累(图4)。在注射1h后,背景器官例如肾脏中的放射性量减少,而肿瘤与背景的比增加。注射24h后,pet扫描显示出非常高的肿瘤摄取,其证实了肿瘤中的富集。
用阻断实验证明了与psma的结合特异性:共注射未标记的psma-617[2mg/kg]导致注射1小时后64cu-ca003在c4-2肿瘤中(30.83±12.61%id/g到2.35±0.38%id/g)和肾脏中(67.04±20.89%id/g至3.47到0.48%id/g)中的积累显著减少。具有过量未标记的psma的64cu-ca003的pet成像(图6c/6d)清楚地证明了生物分布结果。
从动态pet成像中产生的时间活性曲线显示,在注射后1h,肿瘤与肌肉的比为10.5,肿瘤与血液的比为3.0(表s1)。这些曲线表明肾脏的迅速摄取。器官分布研究(图6a)显示,在1h时高的肾脏摄取量(67.04±20.89%id/g)在24h内大部分清除(7.48±8.51%id/g)。相反,高肿瘤摄取值(30.83±12.61%id/g于1hp.i.)几乎保持不变(19.99±6.43%id/g于24hp.i.)。pet成像证实了放射性示踪剂在肿瘤中的强积累(图4)。在注射1h后,背景器官例如肾脏中的放射性量减少,而肿瘤与背景的比增加。注射24h后,pet扫描显示出非常高的肿瘤摄取,其证实了肿瘤中的富集。在长时间点,即注射后45h,维持高的肿瘤积累(图4)。
iv.2.2c)64cu-psma-ca003与64cu-psma-617和64cu-氯化物的体内比较
为了证明psma-ca003的铜配合物的体内稳定性,将64cu-psma-ca003与64cu-psma-617以及64cu-氯化物进行比较(图20)。在c4-2肿瘤异种移植物的小动物pet研究中对化合物进行研究。结果示于表3。从64cu-ca003的动态pet中获得的时间活性曲线在注射1h后显示出高的肿瘤与肝脏比(4.0),而对于64cu-psma-617,肿瘤与肝脏比为0.37(图20/21和表s1/s2)。
表s2在携带c4-2肿瘤异种移植物的balb/cnu/nu小鼠中,从64cu-psma-617小动物pet获得的时间-活性曲线得出的平均标准化摄取值(msuv)。
为了证明进入肿瘤的物质实际上是64cu-ca003而不是游离的64cu,在c4-2荷瘤小鼠中进行64cu-氯化物pet成像(图20/21)后,对相应组织进行均质化、提取和之后的hplc分析。观察到的64cu-氯化物药代动力学不同于64cu-ca003。64cu-氯化物在最大强度投影下的pet成像显示,注射后2h内肿瘤摄取增加。与64cu-ca003相反,64cu-氯化物显示出非常高的肝脏积累(图20/21)。64cu-氯化物在2h时的肿瘤与肝脏比为0.38,而64cu-ca003的肿瘤与肝脏比为6.3。
注射后1h,从68ga-ca028动态pet成像生成的时间活性曲线和平均suv体重值显示肿瘤与肾脏的比为0.78。在2h时,比值增加至3.0(图15和表s1.1),而配体68ga-ca030和68ga-ca029的肿瘤与肾脏的比较低,分别为0.52和0.33(图3、图25和图23)。
表s1.1在携带c4-2肿瘤异种移植物的balb/cnu/nu小鼠中,从68ga-psma-ca028的小动物pet获得的时间-活性曲线得出的平均标准化摄取值(msuv)。
时间-活性曲线显示了示踪剂的快速清除——68ga-ca028显示了高肿瘤积累和高肾脏值。相反,对于68ga-ca027,发现肿瘤积累时肾脏清除更快(图22)。即使68ga-ca030表示比68ga-ca028更高的肾脏摄取值,但是它在所有化合物中显示出最高的肿瘤摄取(图15c)。小动物pet图像表明了68ga-ca027非常快的肾脏清除和低的肿瘤积累。明显地,放射性示踪剂68ga-ca028、68ga-ca029和68ga-ca030显示出高的肿瘤积累(图27b)。68ga-ca029显示出最高的肾脏摄取,然后是68ga-ca030(图27a)。
vii.首个患者的体内研究
vii.1利用64cu-psma-ca003的pet
图7所示前列腺癌患者的pet成像经海德堡大学医院同意,遵循德国法律,并承认赫尔辛基宣言(许可s321/2012)。
首次人体试验是用200mbq的64cu-psma-ca003进行的。第一次pet成像如图7所示,显示了前列腺特异性膜抗原水平高的患者的成像。很明显,患者患有转移期pca,特别是通过多发性淋巴结转移,主要肿瘤位于右肩。
使用新的铜配体后,观察到铜同位素的药代动力学和肿瘤靶向性显著改善。尽管含64cu的psma-617的在体外的标记率较高(>99%),但观察到体内稳定性差,出现高肝脏摄取(cuic,hanyum,hatoria,等人synthesisandevaluationof[(64)cu]psma-617targetedforprostate-specificmembraneantigeninprostatecancer.amjnuclmedmolimaging.2017;7:40-52)。
新的64cu标记的psma配体是靶向psma和显像psma阳性肿瘤病变的有前景的药物,如小动物pet研究、器官分布和首次人体应用的临床前评估所示。
64cu-psma-ca003配体的患者成像表明其在临床研究中成功转化。
vii.2利用203pb-ca012的实验
两名去势抵抗性转移性前列腺癌患者分别在注射258mbq和310mbq203pb-ca012后0.4h、4h、18h、28h和42h接受了平面全身扫描(gehawkeymillennium,1"crystal,me-准直仪,279kev峰+/-10%,8厘米/分钟)。将图像加载到qdose剂量测定软件套件(abx-cro,德累斯顿)中并进行配准。肾脏、肝脏、脾脏、膀胱、唾液腺(左右腮腺和下颌下腺)和几个肿瘤病变以及全身目的区域(roi)在最合适的时间点使用器官相关的最大阈值百分比(15%至65%)进行分割,并传播到所有其他时间点,执行额外的基于器官的自动刚性配准步骤。这些roi用于确定每个器官、肿瘤和全身的时间-活性曲线(tac)。未校正几何平均值图像的第一个时间点(排泄前)用于将roi计数与注射活性(mbq)进行校准。使用已建立的模型假设从静脉血(6个样本/患者)中计算红骨髓tac[shen,s.,meredith,r.f.,duan,j.,macey,d.j.,khazaeli,m.b.,robert,f.,lobuglio,a.f.:improvedpredictionofmyelotoxicityusingapatient-specificimagingdoseestimatefornon–marrow-targeting90y-antibodytherapy.jnuclmed,43:1245-1253,2002.sgouros,g.bonemarrowdosimetryforradioimmunotherapy:theoreticalconsiderations.jnuclmed,34:689-694,1993.]。
从203pb数据中获得的所有tac都使用qdose的置换核素函数进行了重新计算,该函数会自动校正源同位素物理衰变的所有时间点,仅保留其生物清除率;然后应用替换放射性核素的物理衰变。
双指数曲线拟合适用于所有器官tac(少数肿瘤和腺体除外,必须按单指数拟合)。假设从0到第一个测量时间点线性增加,从第一个测量时间点到最后一个测量时间点的数值使用梯形近似,从最后一个测量时间点到无穷大使用拟合函数,来对累积活性进行积分。剩余身体部分是通过从全身减去所有源器官来计算的。在olinda1.1[stabin,m.g.,sparks,r.b.,crowe,e.olinda/exm:thesecond-generationpersonalcomputersoftwareforinternaldoseassessmentinnuclearmedicine.jnuclmed,46(6):1023-1027,2005]中,使用男性-成人-体模的器官质量导出了肾脏、肝脏、脾脏、膀胱内容物、红骨髓和剩余身体部分的停留时间,用于剂量计算。
潜在治疗核素212pb进一步衰变为212bi、212po和208tl。假设子核素保留在母体核素的衰变点以及212pb与其子核素之间的瞬态平衡,对子核素应用了与212pb相同的停留时间。对所有核素分别进行了olinda计算。根据其分支比,使用212po为64.07%、208tl为35.93%的权重因子,对各自的衰变步骤进行了加和。根据医学内照射剂量委员会和美国能源部的建议[sgourosg,roeskejc,mcdevittmr等人.mirdpamphletno.22(abridged):radiobiologyanddosimetryofalpha-particleemittersfortargetedradionuclidetherapy.jnuclmed.2010;51:311–28.feinendegenle,mcclurejj.meetingreport:alpha-emittersformedicaltherapy—workshopoftheunitedstatesdepartmentofenergy,denver,colorado,may30–31,1996.radiatres.1997;148:195–201.],使用α的权重因数为5,β和光子辐射的权重因数为1来使物理吸收剂量被转化为等效剂量;因此,反映关于确定性辐射效应的相对生物功效-被认为是治疗环境中的主导因素。基于ct分割分别测量肿瘤和唾液腺体积,并通过使用内插球形模型估算的幂函数来估算它们的吸收剂量[stabin,m.g.,konijnenberg,m.re-evaluationofabsorbedfractionsforphotonsandelectronsinsmallspheres.jnuclmed,41:149-160,2000.]。
患者的203pb成像数据
注入活性为258mbq-310mbq时,发现从203pb发射的279kevγ射线在80%丰度概率下足以获得清晰的平面扫描(图10)。这些扫描显示,用铅同位素标记的示踪剂在靶组织中特异性富集。因此,该方法提供了将铅同位素用于预期应用的成功方法。
(荷瘤小鼠中203pb-psma-ca012的器官分布在图11的表中显示)
vii.3剂量测定估计203pb-ca012
剂量测定估计
诊断203pb-ca012的剂量测定估计示于图11的表格左列。所有器官吸收的剂量都以光子为主(279kev的初级发射),低概率发射在所有器官中的贡献总和分别<10%。250mbq至300mbq的典型临床检查,例如用于个体剂量测定预测,转化为6.0msv至7.5msv的辐射负荷。
在从212pb衰变至稳定的208pb期间,无论是通过钋还是铊分支,每个原子都会发射两个β粒子和一个α粒子。考虑到完整的后续衰变链,治疗性212pb-ca012的安全剂量测定评估如图11右列所示。假设α的rbe=5,β和γ辐射的rbe=1,治疗性212pb-ca012的等效剂量包括96.4%的α、2.2%的β和1.4%的γ贡献。值得注意的是:由203pb直接追踪的212pb的初始β衰变对总当量剂量的贡献小于1%;99%的当量剂量来自随后的子核素。
在olinda器官中,肾脏和红骨髓以及使用球形模型评估的唾液腺可能有剂量限制。放射性药物的治疗范围由肿瘤剂量与剂量限制器官之间的比来定义。图12总结了最相关的剂量测定信息,并与其他psma靶向α疗法进行了比较。
vii.468ga-ca028和68ga-ca030的人体pet扫描
进行如先前2015年afshar-oromieh等人(afshar-oromieha,hetzheimh,kratochwilc,等人.thetheranosticpsmaligandpsma-617inthediagnosisofprostatecancerbypet/ct:biodistributioninhumans,radiationdosimetry,andfirstevaluationoftumorlesions.jnuclmed.2015;56:1697-1705.)所述的ca028辐射剂量测定的方法。
分别使用68ga-ca028和68ga-ca030通过静脉注射(每个患者339mbq/20纳摩尔68ga-ca028或295mbq/20纳摩尔68ga-ca030)获得图像。在注射1h和3h后的时间点进行68ga-ca028的诊断性检查。(见图14)
在对每个患者分别肘前注射339mbq/20纳摩尔68ga-ca028或295mbq/20纳摩尔68ga-ca030后1h和3h,进行诊断性psma-pet/ct检查。进行如先前afshar-oromieh等人所述的用于评估生物分布的方法(afshar-oromieha,hetzheimh,kratochwilc,etal.thetheranosticpsmaligandpsma-617inthediagnosisofprostatecancerbypet/ct:biodistributioninhumans,radiationdosimetry,andfirstevaluationoftumorlesions.jnuclmed.2015;56:1697-1705)。使用临床标准软件syngo(siemens)测定源器官的活性分布,该软件用于定义pet图像中的voi。该参考文献afshar-oromieh等人也被用作68ga-psma-617的参考标准。
为了证明68ga-ca028和68ga-ca030的临床应用性,对第一个患者进行pet/ct成像。得到的图像如图13a和图13b所示。利用68ga-ca028和68ga-ca030的成像证实了在体外和动物模型中获得的结果。pet扫描和suv是用标准扫描仪设置获得的,对于纯正电子发射器进行了校准(wadastj,pandyadn,solingapuramsaikk,mintza.moleculartargetedalpha-particletherapyforoncologicapplications.ajramjroentgenol.2014;203:253-260)。注射后1h对比3h获得的suv值示于下表。如这些值所反映的,在肿瘤中达到了高且稳定的积累。
表:基于olinda的成年男性模体,68ga-ca028、68ga-psma-617(值来自afshar-oromie等人,2015(9))和68ga-ca030的诊断性suv平均值的安全剂量测定。
- 还没有人留言评论。精彩留言会获得点赞!