由有机材料催化热解制备含氮芳香性化合物的方法与流程
本发明一般地涉及有机材料催化热解制备含氮芳香性化合物的方法,具体地,本发明尤其涉及由生物质催化热解制备吡嗪类化合物、吡啶类化合物、吡咯类化合物、吲哚类化合物、苯胺类化合物或它们的组合的方法。
背景技术:
生物质碳水化合物是自然界存在最多、分布最广的一类重要的有机化合物,主要由碳、氢、氧所组成,葡萄糖、蔗糖、淀粉和纤维素等都属于糖类化合物。随着全球范围内煤和石油资源的消耗和污染问题的日益严重,人们寻找一种可再生的、洁净能源的需求越来越迫切。相比于化石能源,生物质能源具有分布广、总量大、无污染、可再生的特点。此外,废弃蛋白、城市有机垃圾等废弃物大量流失,未得到有效利用,造成极大的资源浪费和环境污染。生物质资源是有机碳的唯一可持续来源,也是唯一可以转化为液体燃料的可再生资源。生物质出发制备高附加值化学品也变得越来越重要。生物质热解尤其是快速热解可以得到高附加值的液体燃料和化学品,被认为是利用生物质的最有效的方式之一。选择性催化热解是在加入催化剂的条件下使生物质定向热解从而提高一种或者几种产物的产率。吡嗪类化合物是1、4位含两个氮杂原子的六元杂环化合物。分子式C4H4N2。两个氮原子占1、2两位的称为哒嗪,占1、3两位的称为嘧啶,它们都是吡嗪的同分异构体。无色晶体。熔点54℃,沸点115~116℃,液态的相对密度1.0311(61/4℃)。具有与吡啶类似的气味。溶于水、乙醇、乙醚等。吡嗪是很弱的碱。它的芳香性与吡啶类似,很不容易发生亲电取代反应,而对亲核试剂比较活泼。碳原子上的氢被甲基或卤素取代后,卤素或甲基上的氢具有活性。吡嗪类化合物可以作为重要的医药中间体和香精香料中间体,也是一种具有高附加值的精细化学品。其衍生物有较多应用,如六氢吡嗪(或称哌嗪)的柠檬酸盐是一个常用的牲畜驱虫剂,对蛔虫特别有效;2-甲基吡嗪是一种重要医药中间体,可以制备抗结核药物吡嗪酰胺;2-甲基吡嗪-5-羧酸可以制备具有降血糖,降血压等药物;2,5-二酮哌嗪是制备肽类化合物的试剂;二苯并吡嗪是一种重要染料,二苯并吡嗪是重要的染料,吩嗪(phenazine)以具有抗肿瘤,抗菌和利尿的性质而著称。而四甲基吡嗪被报道可以捕捉超氧阴离子,并减少人体有粒细胞的氮氧化物产生。吡啶及烷基吡啶是含有一个氮原子的六元环化合物,通常将吡啶及其衍生物统称为吡啶碱类化合物,主要包括吡啶,2-甲基吡啶,3-甲基吡啶,4-甲基吡啶等。吡啶及其衍生物是非常重要的化工中间体,广泛应用于医药、农药、饲料、合成橡胶和印染工业,还可用于表面活性剂和食品添加剂的生产。工业上吡啶主要用于生产磺胺、青霉素、维生素A、可的松、驱虫药和局部麻醉药等,还可用于稳定剂、软化剂、油漆溶剂、合成树脂的缩合剂,以及用于合成除草剂、防腐剂和羟基吡啶等。2-甲基吡啶可用于制造合成橡胶工业的重要原料2-乙基吡啶,亦用于医药工业制氨丙嘧吡啶、长效磺胺、泻药和胶片感光胶添加剂、树脂的原料和燃料中间体等。3-甲基吡啶可用于制维生素B、尼可拉明、强心剂、杀虫剂、防水剂等,其最重要的用途是制造烟酸和烟酰胺,烟酸和烟酰胺可用做饲料工业的添加剂。4-甲基吡啶主要用于有机合成的原料和溶剂,还可用来制取治疗结核药物异烟肼、同时也是制取染料、农药、催化剂、橡胶硫化促进剂和合成树脂的原料。随之吡啶衍生物需求的增长,从炼焦副产品回收分离吡啶及其衍生物的方法难以满足市场需求。Chichibabin提出了以醛和氨为原料大批量生产吡啶及其衍生物的工艺路线,然而所用原料如醛、酮都是大宗化工基本原料,主要从石油化工而来。吡咯类化合物广泛存在于植物、动物和微生物的代谢产物中,吡咯类生物单体是一种重要的五元氮杂环化合物,许多天然和合成的多取代吡咯化合物大多具有抗菌、抗炎和镇痛作用。吡咯类化合物作为精细化工产品的重要中间体在医药、食品、农药、日用化学品、涂料、纺织、印染、造纸、感光材料高分子材料等领域具有广泛用途。如吡咯可以合成具有生物活性的卟吩胆色素原、具有抗菌活性的藤黄绿脓菌素、N-甲基-2-乙酰基吡咯、零红菌素和烧烤香型香精2-乙酰基吡咯等。近年来,吡咯在导电聚合物领域也得到了应用,因而对其需求量日益增加。近年来,吡咯类化合物的工业生产原料主要来自于石油化工产品。吲哚是基本化工原料,广泛用于医药、农药、染料、食品以及香料等众多领域,近年来随着吲哚下游产品的快速发展以及新应用领域的不断衍生,由其衍生出许多重要的高附加值的医药和农药,特别是吲哚衍生物色氨酸的需求增长,使得全球吲哚需求量将大幅增长。吲哚的市场前景十分广阔。目前工业上主要采用以苯胺和乙二醇为原料经过多相催化一步法合成吲哚。苯胺是一种重要的工业化学品,可作为橡胶硫化促进剂、染料、媒染体、药品、炸药原料,和二苯基甲烷聚氨酯(MDI)。烷基取代苯胺,如甲苯胺、枯胺、甲基枯胺、二甲基苯胺、二异丙基苯胺作为原料时,可以增加显影剂、农业试剂和药品的要用。近年来,苯胺用量持续增加,我国从2003年需求量为34万t/a上升到206.5万t/a。全球苯胺用量更多。预计,到2015年,全球苯胺消耗量将达到650万t/a。当前,苯胺的主要生产方法为硝基苯催化加氢法,苯酚氨化法,硝基苯铁粉还原法,其中90%为硝基苯催化加氢法。利用芳族硝基化合物制备方法需要消耗大量硫酸或者硝酸作为芳族化合物的硝化剂,紧接着中和反应需要大量的碱,此外在形成硝基化合物时会产生氧化氮气体,这会引起空气污染。本发明是一种通过对催化剂、反应条件的调控催化热解各种生物质及其衍生物以及农林和城市废弃物选择性的制备吡嗪类、吡啶类,吡咯类,吲哚类以及苯胺类化合物的方法。
技术实现要素:
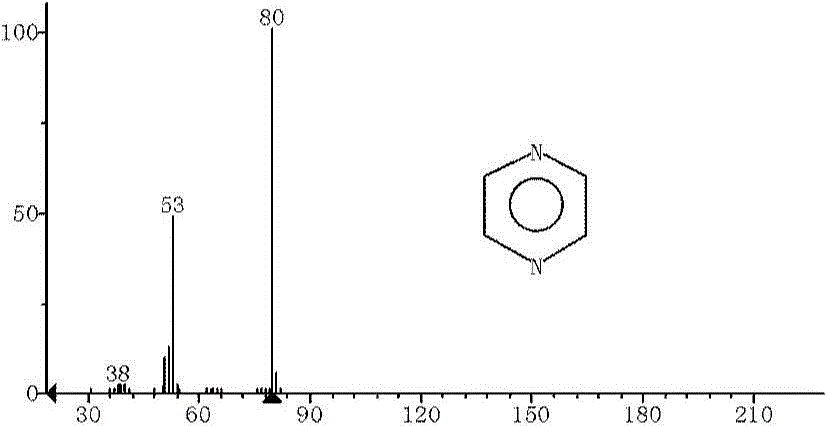
本发明涉及有机材料在含氮气体的存在下的催化热解。在本发明的一个实施方案中,有机材料包括农业和城市固体废物、食品废物、动物废物、碳水化合物、木质素纤维素等,或它们的组合。在本发明的一个实施方案中,有机材料包括木材、甘蔗渣、竹子、玉米秸秆、废弃纸张、油菜籽饼、小桐子饼、饼粕、酒糟、废弃蛋白、微藻或它们的组合。在本发明的一个实施方案中,有机材料包括葡萄糖、纤维二糖、纤维素、淀粉、木糖、木糖醇、木聚糖、壳聚糖、甲壳素、蔗糖、果糖、葡糖糖水溶液、甲基呋喃、2,5-二甲基呋喃、糠醛、5-羟甲基糠醛、5-甲基糠醛、γ-戊内酯、纤维素生物油、水溶性生物油、非水溶性生物油等,或它们的组合。一种由有机材料催化热解制备含氮芳香性化合物的方法,所述方法包括:将有机材料和催化剂进料至反应器;在所述反应器中,在含氮气体的存在下,在加热条件下,使所述有机材料进料在催化剂的催化下进行反应,产生包含一种或多种含氮芳香性化合物的反应体系流。在本发明的一个实施方案中,在所述分离所述反应体系流中含氮芳香性化合物中的至少一部分中,除获得所述包含所分离的含氮芳香性化合物的产物流之外,还获得再循环流;将至少一部分所述再循环流进料至所述反应器。在本发明的一个实施方案中,所述含氮气体包括NH3、有机胺如甲胺、二甲胺、乙胺和/或惰性气体。在本发明的一个实施方案中,所述含氮气体中包含摩尔比大于约1:19的NH3和惰性气体。在本发明的一个实施方案中,所述含氮气体中包含摩尔比为约1:19、1:9、1:4、1:1、4:1、19:1、1:0等的NH3和N2。在本发明的一个实施方案中,所述含氮芳香性化合物包括吡嗪类化合物、吡啶类化合物、吡咯类化合物、吲哚类化合物、苯胺类化合物等,或它们的组合。在本发明的一个实施方案中,吡嗪类化合物包括吡嗪,2-甲基吡嗪,2,5-二甲基吡嗪,2,6-二甲基吡嗪等化合物;吡啶类化合物包括吡啶,2-甲基吡啶,3-甲基吡啶,4-甲基吡啶,以及二甲基吡啶等化合物;吡咯类化合物包括吡咯,2-甲基吡咯,3-甲基吡咯,2,5-二甲基吡咯等化合物;吲哚类化合物包括吲哚,1-甲基吲哚,2-甲基吲哚,3-甲基吲哚,2,8-二甲基吲哚等化合物;苯胺类化合物包括苯胺,邻甲基苯胺,间甲基苯胺,对甲基苯胺,二甲基苯胺等化合物。在本发明的一个实施方案中,催化剂包括沸石催化剂、非沸石催化剂、金属催化剂和/或金属氧化物催化剂等中的一种或多种。在本发明的一个实施方案中,催化剂包括γ-Al2O3、SiO2-Al2O3、WO3/ZrO2、SO42-/ZrO2、MCM-41、ZK-5、ZSM-23、SSZ-20、β-沸石、Y-沸石、ZSM-5和/或HZSM-5等。在本发明的一个实施方案中,催化剂具有15:1至200:1的SiO2/Al2O3,如15:1、25:1、50:1、63:1、80:1、100:1、150:1或200:1等的SiO2/Al2O3。在本发明的一个实施方案中,催化剂与所述有机材料的质量比为1:100至100:1,如1:100、1:50、1:20、1:15、1:10、1:5、1:2、1:1、2:1、5:1、10:1、15:1、20:1、50:1、100:1等。在本发明的一个实施方案中,催化剂与所述有机材料的质量比为1:1至10:1,并且所述含氮芳香性化合物是吡咯类化合物和/或吡嗪类化合物。在本发明的一个实施方案中,催化剂与所述有机材料的质量比为5:1至20:1,并且所述含氮芳香性化合物是吲哚类化合物。在本发明的一个实施方案中,催化剂由多个催化剂颗粒催化,且最大横截面积尺寸小于约1微米的颗粒占催化剂体积总和的至少约50%。在本发明的一个实施方案中,催化剂具有约5埃至约100埃的孔。在本发明的一个实施方案中,所述催化剂包括多个孔;一种或多种催化剂的至少约95%的孔具有在第一尺寸分布和第二尺寸分布内的最小横截面积直径;至少5%的孔在具有在所述第一尺寸分布内的最小横截面积直径;至少约5%的孔具有在所述第二尺寸分布内最小横截面积直径;第一尺寸分布和第二尺寸分布不重叠。在一些实施方式中,可能希望使用一种或多种催化剂以建立双峰孔径分布。在一些情况下,可使用具有双峰孔径分布的单个催化剂。在其他一些情况下,可以使用两种或者多种催化剂的混合物以建立双峰分布。在一些实施方式中,一种或多种催化剂之一包括沸石催化剂,且一种或多种催化剂的另一种包括非沸石催化剂。在一些实施方式中,一种或多种催化剂的至少约70%、至少约80%、至少约90%、至少约95%、至少约98%或至少约99%的孔隙具有在第一尺寸分布或第二尺寸分布内的最小横截面积直径。在一些情况下,一种或多种催化剂的至少约2%、至少约5%或至少约10%的孔隙具有在第一尺寸分布内的最小横截面积直径;且一种或多种催化剂至少约2%、至少约5%或至少约10%的孔隙具有在第二尺寸分布内的最小横截面积直径。在一些情况下,第一尺寸分布和第二尺寸分布选自以上提供的范围。在本发明的一个实施方案中,所述催化剂含有以下掺杂金属中的一种或多种:Cu、Mn、Co、Fe、Ni、Zn、Ga、Pt、In、Ru、Rh、Ir、Pt、Pd、Au、Re、T1和镧系金属等。在本发明的一个实施方案中,通过干/湿法浸渍或离子交换等方式将所述掺杂金属掺杂至所述催化剂中。在本发明的一个实施方案中,催化剂为HZMS-5并且含氮芳香性化合物是吡咯类化合物。在本发明的一个实施方案中,催化剂为SO42-/ZrO2并且含氮芳香性化合物是吡嗪类化合物。在本发明的一个实施方案中,反应器中的反应温度为300℃至800℃,如300℃、400℃、500℃、600℃、700℃、800℃等。在本发明的一个实施方案中,反应器中的反应温度为300℃至500℃,并且含氮芳香性化合物是吡咯类化合物。在本发明的一个实施方案中,反应器中的反应温度为500℃至800℃,优选600℃,并且含氮芳香性化合物是吲哚类化合物和/或苯胺类化合物。在本发明的一个实施方案中,所述反应器是固定床反应器和/或流化床反应器等。在本发明的一个实施方案中,所述反应器是循环流化床反应器和/或湍动流化床反应器等。在本发明的一个实施方案中,以0.05至2的WHSV(质量归一化),如0.05、0.1、0.2、0.5、0.8、1、1.5、2的WHSV将所述有机材料进料至所述反应器中。在本发明的一个实施方案中,以0.8至2的WHSV(质量归一化)将所述有机材料进料至所述反应器中,并且含氮芳香性化合物是吡咯类化合物。在本发明的一个实施方案中,以0.1至0.8的WHSV(质量归一化)将所述有机材料进料至所述反应器中,并且含氮芳香性化合物是吲哚类化合物和/或苯胺类化合物。本发明首次开发了通过对催化剂种类、用量、反应温度等因素的调控采用催化热解方法有选择性地转化生物质、生物油以及生物质基化学品到吡嗪类、吡咯类、吡啶类、吲哚类以及苯胺类化学品等石油化工中的大宗和精细化学品的新线路。与现有技术的方法相比较,本发明主要是克服了以前生产这些化学品的工艺中的以下缺点:如:(1)本发明原料全部是可再生资源涵盖所有生物质材料,之前产物原料来源于石油化工产品;(2)本发明的生产工艺是绿色的生产工艺;不会有之前生产过程中需要大量酸碱,有环境污染等缺点;(3)以生物油或者生物质基化学品为原料时,这条线路可与石油化工中反应线路相近或者完全可以替代;(4)本线路由原料到生产工艺全过程是一个可再生、绿色、环保线路。附图说明图1是催化剂热解制备吡嗪类和吡咯类化合物气质图及质谱图。图2是催化热解制备吲哚类化合物气质图及质谱图。图3是催化热解制备苯胺类吲哚类化合物气质图及质谱图。图4是催化热解制备吡啶类和吡咯类化合物气质图及质谱图。图5是实施例1所示的反应模型流程图。图6是催化热解水溶性生物油(实施例14)反应流程示意图。图7是本发明的一个实施方案的工艺流程图,其中1系列为设备;1-1:干燥器;1-2:研磨系统;1-3:反应器;1-4:产物反应器;1-5:固体分离器;1-6:冷凝系统;1-7:蒸汽回收系统;1-8:催化剂再生器;1-9:压缩系统;1-10:流体源。2系列为流体线路图;2-1:进料组合物,包括生物质、催化剂和载气;可以为固体、液体和/或气体;2-2:水分;2-3:反应产物流;2-4:分离催化剂后产物流;2-5:分离的催化剂流;2-6:氧化剂,可包括氧气,空气,水蒸气;2-7:再生后催化剂流;2-8:催化剂再循环流;2-9:产物反应后产物流;2-10:分离器吹扫流;2-11:催化剂再生器吹扫流;2-12:再生器排出流;2-13:产物液体组分流;2-14:产物气体组分流;2-15:蒸汽流;2-16:CO、CO2或其他不可回收气体蒸汽流;2-17:其他产物,如额外气体。具体实施方式本说明书公开了用于制备诸如吡嗪类、吡啶类、吡咯类、吲哚类和苯胺类等含氮芳香类的化学品的组合物和方法,更具体地,通过催化热解制备化学品的组合物和方法。一些实施方式涉及通过催化热解工艺(例如,催化快速热解)制备流体(例如,液体、超临界流体和/或气体)产物,例如含氮芳香类化学品(例如,吡嗪、吡咯、吲哚、苯胺等)的方法。在一些实施方式中,产物或其一部分在标准环境温度和压力(SATP,即25℃和100kPa绝对压力)下为液体。一些这样的方法可包括使用包含有机材料(例如,液体、气体和/或固体有机材料)和非均相热解催化剂组分的混合物的组合物。在一些实施方式中,可将含有机材料进料至反应器,进行催化热解,并且可将一部分产物流再循环至包含有机材料的进料流。在本发明的一些实施方案中可以产生的产物如表1所示。表1.本发明的产物中文名称与沸点化合物名称沸点(℃)化合物名称沸点(℃)化合物名称沸点(℃)化合物名称沸点(℃)吡咯131吲哚253-2542-甲基吡咯147吲嗪-3-甲基吡咯144N-甲基吲哚2712,5-二甲基吡咯-甲基吲哚-吡啶115.32-甲基吲哚2732-甲基吡啶128-1293-甲基吲哚265-2663-甲基吡啶1442,8-二甲基吲哚-4-甲基吡啶1453,8-二甲基吲哚-吡嗪115-116苯胺1842-甲基吡嗪1352-甲基苯胺199-2002,5-二甲基吡嗪1553-甲基苯胺203-2042,6-二甲基吡嗪1544-甲基苯胺200在一些实施方式中,所述混合物可在高温(例如,300℃至800℃)下热解。热解过程至少需要足以产生部分可分离和可识别的含氮芳香产物流体的时间。本文所述的方法还可包括使用专用催化剂。例如,在一些情况下,使用沸石催化剂;本文所用的催化剂可任选具有任意的二氧化硅与氧化铝摩尔比。在一些情况下,催化剂可以有相对小的颗粒形成或包括相对小的颗粒,所述颗粒可以结块。在一些情况下,进料至热解反应器的组合物具有任意的催化剂与有机材料质量比。一些实施方式可涉及生物质的单级热解法。此类方法可包括提供或使用单级热解装置。单级热解装置是在单个容器中进行热解和随后的催化反应的装置。在一些实施方式中,单级热解装置包括流化床反应器。如下文更详细地描述的那样,多级装置也可用于制备流体含氮芳香性化学品。本文所用的术语“热解”是其在本领域中的常规含义,用于表示在不氧化的情况下仅通过热使化合物(例如有机材料)转化成一种或多种其他物质(例如挥发性有机化合物、气体和焦炭),这可在使用或不使用催化剂的情况下进行。“催化热解”是指在催化剂存在下进行的热解,可包括下文中更详细地描述的步骤。例如,在Huber,G.W.等,“SynthesisofTransportationFuelsfromBiomass:Chemistry,Catalysts,andEngineering”Chem.Rev.106,(2006),第4044-4098页中概括了催化热解工艺的实例,其通过引用整体并入本文。本文所用的术语“生物质”是其在本领域中的常规含义,用于表示能量或化学品的任何可再生的有机来源。其主要组分可为(1)树(木材)和所有其他植物;(2)农产品与废弃物;(3)藻类和其他海洋植物;(4)代谢废物(肥料、下水道污物);和(5)纤维质城市废物。例如在Huber,G.W.等,“SynthesisofTransportationFuelsfromBiomass:Chemistry,Catalysts,andEngineering”Chem.Rev.106,(2006),第4044-4098页中描述了生物质材料的实例。发明人发现,对于某些反应,反应条件的某些变化和此类变化的组合可产生有利的产物和/或产率、更低的焦炭形成产率和/或更受控的产物形成。例如,应用高温(例如,在反应器和/或固体分离器中)可由在较低温度下不会发生的反应产生有利的产物和/或产率。本发明人还发现,提供具有高的催化剂与有机材料质量比的进料可产生吲哚类和苯胺类化合物产物的理想产率。例如,不希望受理论限制,本发明人相信,高加热速率和高催化剂与进料质量比可有利于将由有机进料热解形成的挥发性有机化合物在其热分解前引入到催化剂中,由此导致含氮芳香性化合物的高产率。较低的质量归一化空速也显示产生含氮芳香化合物的理想产率。本发明人还发现,有机材料在系统的高温组件(例如,反应器和/或固体分离器)中的较长停留时间可允许充足时间来进行另外的化学反应以形成期望的产物。在本发明中,本发明人还发现,使用具有特定性质的催化剂有助于形成较大量的含氮芳香性化合物产物。例如,在一些情况下,使用包含较小尺寸的颗粒的催化剂可导致产生较高量的含氮芳香性化合物和/或较低量的焦炭。再例如,在一些实施方式中,发现ZSM-5优先产生含氮芳香性化合物。另外,发现包括Bronstead酸位点和/或良序孔隙结构的某些催化剂在一些情况下选择性地产生含氮芳香性化合物。在一些情况下,催化剂孔径可用于影响所产生的产物化合物的量和类型。本文所述的实施方式还包括用于进行催化热解的化学工艺设计。在一些情况下,所述工艺可包括使用一个或多个流化床反应器(例如,循环流化床反应器、湍动流化床反应器、鼓泡流化床反应器等)。本文所述的工艺设计可任选包括进料到一个或多个反应器中的材料的专门操作。例如,在一些实施方式中,可在将材料供应到反应器之前将该进料材料干燥、冷却和/或研磨。本发明的其他方面涉及应用本文所述的工艺设计制成的产物组合物。不受整个热/催化转化工艺的特定工作模式或步骤顺序的限制,本发明人认为催化热解包括有机材料(例如,诸如固体生物质)部分热解以产生一种或多种热解产物(例如,挥发性有机物、气体、固体焦炭等)和至少使得一部分所述产物流体中一种或多种的热解产物在催化剂条件下进行催化剂催化反应。除一氧化碳、二氧化碳、水和焦炭外,该催化反应还可涉及进入催化剂(例如沸石催化剂)的挥发性有机物,在此将它们转化为化学品(例如含氮芳香性化合物和烯烃)。在催化剂内或在与催化剂接触时,生物质衍生物质可发生一系列脱水、氨化、脱羰、脱羧、异构化和脱氢反应,以生成含氮芳香性化合物、烯烃、CO、CO2和水。选择性制备含氮芳香性化合物的挑战在于能够使碳收率最大化。利用本文所述的方法和工艺可解决燃料生产的问题。例如,通过控制多个工艺参数可由有机材料进料可控地制备含氮芳香性化合物,所述工艺参数尤其包括例如:催化剂选择(包括类型和物理性质例如孔径、粒径、结块的存在和程度、颗粒/结块形状等)、有机材料选择、再循环流组成、反应温度、催化剂与有机材料质量比(例如,在进料流中、在反应器中等)、催化剂二氧化硅与氧化铝摩尔比、质量归一化空速、在各种加工组件中的停留时间。可选择工艺参数以使焦炭形成速率较低。在一些实施方式中包括有机材料的反应的化学工艺。在一些实施方式中,该方法可包括在足以产生一种或多种热解产物的反应条件下在反应器(例如,流化床反应器)中热解至少一部分有机材料。此外,该方法可包括在足以产生一种或多种流体产物的反应条件下用催化剂使至少一部分所述一种或多种热解产物催化反应。在一些实施方式中,可由所述热解产物通过脱水、氨化、脱羰、脱羧、异构化和脱氢反应制备一种或多种流体产物;该热解和催化反应方法可在单个反应器中进行。在一些情况下,该化学方法可用于生产特定流体产物(例如含氮芳香性化合物和/或烯烃);并且,可将由该化学方法产生的一部分烯烃再循环至进料流,通过该进料流,有机材料被进料至反应器(例如,热解反应器)。图7包括根据一组实施方式用于进行催化热解的示例性化学方法设计的示意图。在一些实施方式中,这种方法可用于进行催化热解。如图7的示例性实施方式所示,进料流2-1包括待进料至反应器1-3的有机材料的进料组合物。有机材料通常可包含碳、氢、氧(其中碳是按质量计最多的组分)以及次要比例的其他元素,如氮和硫。该进料组合物中的有机材料可包括固体、液体和/或气体。下文提供有机材料的具体实例。在一些实施方式中,进料组合物(例如,在图7的进料流2-1中)包括有机材料和催化剂的混合物。混合物可包括例如固体、液体和/或气体。在一些实施方式中,混合物包括固体催化剂和固体有机材料的组合物。在其他实施方式中,可与进料组合物分开提供催化剂。如下文更详细地描述的,可使用多种催化剂,例如可使用具有不同的二氧化硅与氧化铝摩尔比和/或不同的孔径的沸石催化剂。在一些实施方式中,例如当使用固体有机材料时,可任选在将其进料到反应器之前例如通过任选的干燥器1-1从进料组合物中除去水分2-2。出于几个原因,从进料流中除去水分是有利的,例如,进料流中的水分可能需要额外的能量输入才能将该进料加热至足够高的温度以实现热解;进料的水分含量的变化会在控制反应器温度方面造成困难;另外,从进料中除去水分可减少或省去在后继的加工步骤中处理水的需要。在一些实施方式中,可以干燥进料组合物直至该进料组合物包含少于约10重量%,少于约5重量%,少于约2重量%或少于约1重量%的水。能够从进料组合物中除去水的合适设备是本领域技术人员已知的。例如,在一组实施方式中,干燥器包括被加热至特定温度(例如,至少约80℃,至少约100℃,至少约150℃或更高)的炉,进料组合物连续、半连续或周期性地通过该炉。在一些情况下,干燥器可包括真空室,进料组合物分批进入其中处理。干燥器的其他实施方式可将高温与真空操作结合。干燥器可一体连接到反应器上或可作为与反应器分开的单元来设置。在一些情况下,在进料进入反应器之前,可在任选的研磨系统1-2中降低进料组合物的粒度。在一些实施方式中,离开研磨系统的经磨碎的进料组合物的平均直径可为进入该研磨系统的进料组合物的平均直径的不大于约50%、不大于约25%、不大于约10%、不大于约5%、不大于约2%。大颗粒进料材料比小颗粒进料材料更容易运输。在一些情况下,将小颗粒进料到反应器中是有利的。研磨系统的使用可允许在来源与生产工艺之间进行大颗粒进料,同时能够将小颗粒进料到反应器中。能够研磨进料组合物的合适的设备是本领域技术人员已知的。例如,研磨系统可包括工业磨机(例如,锤磨机、球磨机等)、具有刀片的单元(例如切片机、切碎机等)或任何其他合适类型的研磨系统。在一些实施方式中,研磨系统可包括冷却系统(例如主动冷却系统,如泵送流体热交换器,被动冷却系统,如包括翅片的系统,等等),其可用于在将进料组合物引入反应器之前使该进料组合物保持在较低的温度(例如环境温度)。研磨系统可一体连接到反应器上或可作为与反应器分开的单元设置。虽然在图7中研磨步骤显示为在干燥步骤后,但在一些实施方式中可颠倒这些操作的顺序。在其他实施方式中,干燥和研磨步骤可使用集成单元来实现。在一些情况下,可使用分开的单元来实现有机材料的研磨和冷却。有机材料的冷却可能是合乎需要的,例如,以减少或防止进料材料在进入反应器之前的不想要的分解。在一组实施方式中,可将有机材料进料到研磨系统以制备磨碎的有机材料。该磨碎的有机材料可随后从该研磨系统进料至冷却系统并冷却。可在将有机材料引入反应器之前将该有机材料冷却至低于约300℃、低于约200℃、低于约100℃、低于约75℃、低于约50℃、低于约35℃或低于约20℃的温度。在包括使用冷却系统的实施方式中,该冷却系统包括能够降低生物质温度的主动冷却单元(例如热变换器)。在一些实施方式中,干燥器、研磨系统和冷却系统中的两个或多个可以组合在单个单元中。在一些实施方式中,冷却系统可与一个或多个反应器直接合为一体。如图7中所示,可将进料组合物转移至反应器1-3中。在一些情况下,该反应器可用于实施有机材料的催化热解。在图7的示例性实施方式中,反应器包括本领域技术人员已知的任何合适的反应器。例如,在一些情况下,反应器可包括连续搅拌釜反应器(CSTR)、间歇式反应器、半间歇式反应器或固定床催化反应器。在一些情况下,反应器包括流化床反应器和固定床反应器,例如循环流化床反应器。在一些情况下,流化床反应器可改进在热解和/或后继反应过程中催化剂和/或有机材料的混合,这可增强对形成的反应产物的控制。流化床反应器的使用也可改进反应器内的热传递。另外,在一些情况下,流化床反应器中改进的混合可导致附着于催化剂上的焦炭量降低,从而降低催化剂的失活。本文所使用的术语“流化床反应器”是其在本领域中的常规含义,用于指包括可容纳粒状固体材料(例如二氧化硅颗粒、催化剂颗粒等)的容器的反应器,其中流体(例如气体或液体)以高至足以悬浮该固体材料并使其表现得像流体的速度通过该粒状固体材料。术语“循环流化床反应器”也是其在本领域中的常规含义,用于指如下的流化床反应器:其中粒状固体材料离开该反应器,通过与该反应器流体连同的管线循环并再循环回该反应器中。鼓泡流化床反应器和湍动流化床反应器也是本领域技术人员已知的。在鼓泡流化床反应器中,对于用于使粒状固体材料流化的流体流在足够低的流速下操作,使得在操作过程中在该流化床的容积内观察到气泡和空隙。在湍动流化床反应器中,流化流的流速高于鼓泡流化床反应器中所采用的流速,因此,在操作过程中,于该流化床的容积内没有观察到气泡和空隙。本文所使用术语“固定床反应器”是尤其在本领域中常规定义,又称填充床反应器,装填有固体催化剂或固体反应物用以实现多相反应过程的一种反应器。固体物通常呈颗粒状,粒径2~15mm左右,堆积成一定高度(或厚度)的床层。床层静止不动,流体通过床层进行反应。它与流化床反应器及移动床反应器的区别在于固体颗粒处于静止状态。涓流床反应器也可归属于固定床反应器,气、液相并流向下通过床层,呈气液固相接触。固定床反应器有三种基本形式:①轴向绝热式固定床反应器流体沿轴向自上而下流经床层,床层同外界无热交换。②径向绝热式固定床反应器。流体沿径向流过床层,可采用离心流动或向心流动,床层同外界无热交换。③列管式固定床反应器由多根反应管并联构成。管内或管间置催化剂,载热体流经管间或管内进行加热或冷却,管径通常在25~50mm之间,管数可多达上万根。反应器可具有任何适用于实施本文所述的方法的尺寸。例如,反应器可具有0.1-1L、1-50L、50-100L、100-250L、250-500L、500-1000L、1000-5000L、5000-10,000L或10,000-50,000L的容积。在一些情况下,反应器具有大于约1L的容积,或在其他情况下,具有大于约10L、50L、100L、250L、500L,1,000L或10,000L的容积。大于约50,000L的反应器容积也是可能的。反应器可以是圆筒形、球形或任何其他合适的形状。本发明人发现,当在本文所述的方法和系统中执行反应条件和系统组件的特定组合时,可实现所需产物生产的更高产率、更低焦炭形成产率、和/或更受控地获得目标产物(例如含氮芳香性化合物)。例如,可如下文所述控制反应条件,例如反应器和/或固体分离器的温度、反应器压力、进料流的加热速率、催化剂与有机材料质量比、质量归一化空速、有机材料在反应器中的停留时间、反应产物在固体分离器中的停留时间、和/或催化剂类型(以及沸石催化剂的二氧化硅与氧化铝摩尔比)中的至少一种,以实现有益结果。可在任何合适的温度下操作反应器。在一些情况下,在较高的温度下操作反应器可能是合乎需要的。例如,可在至少约300℃、至少约400℃、至少约500℃、至少约600℃、至少约700℃、至少约800℃、。在一些实施方式中,可在约500℃至约800℃、约525℃至约800℃、约550℃至约700℃或约575℃至约650℃的温度下操作反应器。在低温条件下反应有利于产生吡咯类吡嗪类化合物;在高温条件下,反应剧烈,有利于产生吡啶类、吲哚类以及苯胺类化合物。在其他实施方式中,可在约500℃至约600℃操作反应器。也可在任何合适的压力下操作反应器。在一些实施方式中,可在约0.1-4Mpa的压力下操作反应器。在一些实施方式中,可在至少约1atm、至少约5atm、至少约1Mpa或至少约4Mpa的压力下操作反应器。在一些实施方式中,可选择有机材料的质量归一化空速以选择性地制备所需的一批流体产物。本文所用的术语“质量归一化空速”定义为进入反应器的有机材料的质量流速(例如以克/小时为单位测得)除以该反应器中催化剂的质量(例如以克为单位测得),且单位为时间的倒数。可根据所用反应器的类型应用不同方法计算有机材料在反应器中的质量归一化空速。例如,对于采用间歇式或半间歇式反应器的系统,有机材料没有质量归一化空速。对于在反应过程中将催化剂进料到反应器和/或从反应器中取出催化剂的系统(例如,循环流化床反应器),可通过在操作(例如稳态操作)期间计算反应器容积内催化剂的平均量来测定质量归一化空速。在本文所述的实施方式中可应用任何合适的质量归一化空速。在一些情况下,可应用小于约10小时-1、小于约5小时-1、小于约1小时-1、小于约0.5小时-1、小于约0.1小时-1、小于约0.05小时-1或小于约0.01小时-1的质量归一化空速。在一些实施方式中,可应用约0.01小时-1至约10小时-1、约0.01小时-1至约5小时-1、约0.01小时-1至约0.1小时-1、约0.1小时-1至约1小时-1或约1小时-1至约10小时-1的质量归一化空速。在一些实施方式中,使用流化床反应器采用小于约1小时-1、小于约0.5小时-1、小于约0.1小时-1、小于约0.05小时-1、小于约0.01小时-1、约0.01小时-1至约0.1小时-1或约0.1小时-1至约1小时-1的质量归一化空速也是有利的。一些实施方式包括改变有机材料的质量归一化空速,以选择性地制备不同的流体产物。例如,在一些实施方式中,改变有机材料的质量归一化空速可控制反应产物中含氮芳香性化合物和烯烃化合物的相对量。例如,较低的质量归一化空速可用于制备的含氮芳香性化合物量相对大于烯烃量。较高的质量归一化空速可用于制备的烯烃量相对大于含氮芳香性化合物量。在一些实施方式中,固体有机材料在流化床反应器中以约0.1小时-1至约10小时-1的质量归一化空速提供以选择性地制备烯烃化合物,或以约0.01小时-1至约0.1小时-1的质量归一化空速提供以选择性地制备含氮芳香性化合物。在一些情况下,控制有机材料(例如,固体有机材料)在反应器中和/或在指定的一组反应条件(即该有机材料在给定的反应器系统中发生热解的条件)下的停留时间是有益的。在连续流系统中,有机材料在反应器中的停留时间定义为有机材料和由其形成的任何反应产物(不包括积聚在反应器中的产物,例如沉积在催化剂上的焦炭)在该反应器中度过的时间量。可根据所用反应器的类型应用不同方法计算有机材料在反应器中的停留时间。例如,在反应器包括填充床反应器的实施方式中,仅连续向该填充床反应器中供应有机材料(即不使用载体或流化流),可通过反应器容积除以离开该反应器的产物气体的体积流量测定有机材料在此处所用反应器中的停留时间。在操作过程中对质量流封闭的反应器(例如,间歇式反应器)中发生反应的情况下,有机材料在这类反应器中的停留时间被定义为在容纳该有机材料的反应器中的温度达到足以开始热解反应的水平(例如,对许多典型的有机进料原材料而言,通常约300℃至约800℃)时与在使反应器骤冷(例如冷却至低于足以支持另外的热解的温度,例如对许多典型的有机进料原材料而言,通常约300℃至约800℃)时之间经过的时间量。在一些情况下,例如对某些流化床反应器而言,进料流可包含流化流体。在使用循环流化床的情况下,催化剂和流化流体都可进料/再循环至反应器中。在某些情况下,辅助材料可包含夹带在有机材料中的污染物。在这样的情况下,如上述填充床的情况那样,可通过反应器容积除以离开该反应器的有机材料和反应产物气体的体积流量来测定该有机材料在反应器中的停留时间;然而,由于可能不方便直接测定离开该反应器的有机材料和反应产物气体的流量,因此可通过从离开该反应器的气流的总体积流量中减去进入该反应器的辅助材料(例如流化流体、催化剂、污染物等)的进料体积流量来估算离开该反应器的有机材料和反应产物气体的体积流量。在一些实施方式中,材料(例如,有机材料或任何其他合适的进料材料)在反应器中的停留时间为至少约2秒,至少约5秒,至少约10秒,至少约30秒,至少约60秒,至少约120秒,至少约240秒,或至少约480秒。在一些情况下,材料(例如有机材料或任何其他合适的进料材料)在反应器中的停留时间为小于约5分钟,约1分钟至约4分钟,或约2秒至约480秒。在许多情况下,之前的“快速热解”研究采用具有很短的进料材料(例如,有机材料)停留时间(例如,小于2秒)的系统。但是,本发明人发现,在一些情况下,应用较长的停留时间能够留出充足时间用于附加的化学反应而形成希望的产物。例如,可通过提高反应器的容积和/或降低有机材料的体积流量来实现长的停留时间。但是,应该理解,在本文所述的一些实施方式中,进料材料(例如,有机材料)的停留时间可较短,例如,小于约2秒或小于约1秒。在使用流化床反应器的某些情况下,可通过使流体流过反应器来流化该反应器中的进料材料(例如,固体有机材料)。在图7的示例性实施方式中,使用流体流2-8来流化反应器1-3中的进料材料。可由流体源1-10和/或由反应器的产物流经压缩机1-9(下面将更详细地描述)向流体流供应流体。本文所用的术语“流体”是指通常为液态、超临界态或气态的物质。但是,流体也可含有固体,例如,悬浮或胶体颗粒。在一些实施方式中,控制流化流体在反应器中的停留时间可能是有利的。流化流体的停留时间被定义为反应器的容积除以流化流体的体积流量。在一些情况下,流化流体的停留时间可为至少约5秒,至少约10秒,至少约30秒,至少约60秒,至少约120秒,至少约240秒,或至少约480秒。在一些情况下,流化流体的停留时间可为约2秒至约480秒,约5秒至约480秒,约10秒至约480秒,约30秒至约480秒,约60秒至约480秒,约120秒至约480秒,或约240秒至约480秒。本发明中可用的合适的流化流体尤其包括,例如惰性气体(如氦气、氩气、氖气、氮气等)和氨气。如图7的示例性实施方式所示,在有机材料反应过程中形成的产物(例如,流体产物)经产物流2-3离开反应器。在一些情况下,除反应产物外,产物流可包含未反应的有机材料、流化流体和/或催化剂。在一组实施方式中,可从反应器的流出物流中回收所需的反应产物(例如,液体含氮芳香性化合物、烯烃、气体产物等)。在一些实施方式中,可将任选的产物反应器1-4并入工艺中。例如,可使用产物反应器,以将产物流2-3中的一种或多种流体产物(例如,烯烃、含氮芳香化合物等)转化为一种或多种其他产物。在一些情况下,产物反应器可含有能够用于进行一个或多个催化反应的催化剂(例如,沸石催化剂)。本领域普通技术人员能够选择实施这些反应的合适反应器类型和/或条件。如图7的示例性实施方式所示,可将产物流2-3送入任选的固体分离器1-5。在一些情况下,该固体分离器可用于从产物流中存在的催化剂(例如,至少部分失活的催化剂)分离反应产物。另外,在一些情况下,该固体分离器可用于从催化剂中除去焦炭和/或灰分。在一些实施方式中,该固体分离器可包含任选的吹扫流2-10,其可用于从该固体分离器中吹扫焦炭、灰分和/或催化剂。本领域普通技术人员可容易地设计实现固体分离和/或除焦步骤所需的设备。例如,在一组实施方式中,固体分离器可包括含有筛网材料的容器,该筛网材料界定该容器的保留部分和渗透部分。该筛网可用于将催化剂留在保留部分内,同时使反应产物穿过到达渗透部分。催化剂可通过筛网保留侧上的开口离开固体分离器,而反应产物可离开筛网渗透侧上的开口。可在任何合适的温度下操作固体分离器。在一些实施方式中,可在高温下操作固体分离器。本发明人发现,对于某些反应,在固体分离器中应用高温可实现来自反应器的化合物的另外的重整和/或反应。这可增加期望产物的形成。不希望受任何理论限制,固体分离器中的高温可提供足以驱动吸热重整反应的能量。可在例如约25℃至约200℃、约200℃至约500℃、约500℃至约600℃、或约600℃至约800℃的温度下操作固体分离器。在一些情况下,可在至少约500℃、至少约600℃、至少700℃、至少800℃或更高温度下操作固体分离器。在一些情况下,控制催化剂在固体分离器中的停留时间可为有利的。催化剂在固体分离器中的停留时间被定义为固体分离器的容积除以催化剂通过该固体分离器的体积流量。在一些情况下,期望催化剂在固体分离器中的较长停留时间,以利于从催化剂中除去充足量的灰分、焦炭和/或其他不想要的产物。另外,本发明人发现,通过应用催化剂在固体分离器中的较长停留时间,可使热解产物进一步反应以产生期望产物。在一些实施方式中,一起选择固体分离器中的停留时间和温度以制备所需的产物流。在一些实施方式中,催化剂在固体分离器中的停留时间为至少约1秒、至少约5秒、至少约10秒、至少约30秒、至少约60秒、至少约120秒、至少约240秒、至少约300秒、至少约600秒、或至少约1200秒。控制催化剂在固体分离器中的停留时间的方法是本领域技术人员已知的。例如,在一些情况下,固体分离器的内壁可包括用于约束通过该固体分离器的催化剂流和/或提高流体流在该固体分离器中的路径长度的挡板。另外,可通过控制催化剂通过固体分离器的流量(例如通过控制流化流体通过反应器的流量)来控制催化剂在固体分离器中的停留时间。固体分离器可具有任何合适的尺寸。例如,固体分离器可具有0.1-1L、1-50L、50-100L、100-250L、250-500L、500-1000L、1000-5000L、5000-10,000L或10,000-50,000L的容积。在一些情况下,固体分离器具有大于约1L的容积,或者在其他情况下,固体分离器具有大于约10L、50L、100L、250L、500L、1,000L或10,000L的容积。大于50,000L的固体分离器容积也是可能的。固体分离器可是圆筒形、球形或任何其他形状,并可为循环或非循环的。在一些实施方式中,固体分离器可包括与该工艺中所用的一个或多个反应器相似的容器或其他单元操作。固体分离器中的催化剂流可包含任何合适的几何形状。例如,流径可为基本上直的。在一些情况下,固体分离器可包含具有蜿蜒、曲折、螺旋或任何其他合适形状的流道。固体分离器的流径的长度(或在一些实施方式中,催化剂通过固体分离器的路径长度)与固体分离器通道的平均直径之比可包括任何合适的比率。在一些情况下,该比率可为至少2:1、至少5:1、至少10:1、至少50:1、至少100:1或更大。上述参数可以以任何合适的组合使用,以产生期望的反应产物(例如,含氮芳香性化合物)和/或有利的产率或特定组分。例如,长的停留时间可与循环或湍动流化床反应器联用以加工固体有机材料。在一些实施方式中,在反应器中热解固体有机材料之后,可在固体分离器中应用较高的温度(例如,至少500℃)和长的停留时间(例如,至少约1秒、至少约5秒、至少约10秒、至少约30秒、至少约60秒、至少约120秒、至少约240秒、至少约300秒、至少约600秒、或至少约1200秒等)。在其他实施方式中,较低的质量归一化空速(例如,小于约0.1小时-1、小于约0.05小时-1、小于约0.01小时-1等)可用于在流化床反应器中制备大于烯烃量的含氮芳香性化合物量,例如至少约6%含氮芳香性化合物或更多。在另一组实施方式中,固体有机材料和包括大的二氧化硅与氧化铝摩尔比(例如,至少约15)的沸石催化剂可在反应器中以高速率加热。在一些情况下,可将催化剂和固体有机材料以至少约0.5:1的质量比送入反应器中并加热至例如500℃至800℃的温度。在一些情况下,可将催化剂和固体有机材料以至少约0.5:1的质量比送入反应器中,以使该混合物具有较长的停留时间(例如,至少约5秒)。在又一组实施方式中,可应用较长的流化流体停留时间(例如,至少约5秒)和较高的反应器温度(例如,约500℃至约800℃)。如之前所述,并非在所有实施方式中都需要固体分离器。例如,对于应用催化固定床反应器的情况,可将催化剂留在反应器内,并且反应产物在离开反应器时可基本不含催化剂,因此不需要单独的分离步骤。在图7中所示的实施方式组中,分离的催化剂可经流2-4离开固体分离器。在一些情况下,离开分离器的催化剂可为至少部分失活的。在一些实施方式中,分离的催化剂可被送入催化剂再生器1-8,在此可将任何至少部分失活的催化剂再活化。在一些实施方式中,催化剂再生器可包括任选的吹扫流2-11,其可用于从该再生器中吹扫焦炭、灰分和/或催化剂。活化催化剂的方法是本领域技术人员公知的。在一组实施方式中,例如如图7中所示,将氧化剂经流2-6送入催化剂再生器。该氧化剂可源自任何来源,尤其包括例如氧罐、大气、水蒸气。在再生器中,通过使催化剂与氧化剂反应,再活化该催化剂。在一些情况下,失活的催化剂可包括残留碳和/或焦炭,它们可通过在再生器中与氧化剂反应而除去。图7中的再生器包括排出流2-12,其可包括再生反应产物、残留氧化剂等。催化剂再生器可具有上文关于反应器或固体分离器提到的任何合适的尺寸。另外,在一些情况下,可在高温(例如,至少约300℃、400℃、500℃、600℃、700℃、800℃或更高)下操作再生器。也可利用本领域技术人员已知的方法(包括上述那些方法)控制催化剂在再生器中的停留时间。在一些情况下,将催化剂通过再生器的质量流速与在反应器和/或固体分离器中的流速相联系,以保持系统中的质量平衡。如图7的示例性实施方式中所示,再生的催化剂可经流2-7离开再生器。再生的催化剂可经再循环流2-8再循环回反应器。在一些情况下,催化剂可在操作过程中从系统流失。在一些这样的和其他情况下,可经补充流2-12向系统中添加另外的催化剂。如图7中示例性显示的,再生和补充的催化剂可与流化流体一起经再循环流2-8送入反应器,但在其他实施方式中,催化剂和流化流体可经分开的流送入反应器。再回到图7中的固体分离器1-5,反应产物(例如,流体含氮芳香性产物)经流2-3离开该固体分离器。在一些情况下,可将流2-7中的反应产物送入任选的冷凝器1-6。该冷凝器可包括热交换器,其使至少一部分反应产物从气态冷凝成液态。冷凝器可用于将反应产物分离成气体、液体和固体组分。冷凝器的操作是本领域技术人员公知的。在一些实施方式中,冷凝器也可利用压力变化来冷凝产物流的一部分。在图7中,流2-13可包括反应产物的液体组分(例如,水、含氮芳香性化合物、烯烃化合物等),且流2-14可包括反应产物的气体组分(例如,CO、CO2、H2,NH3等)。在一些实施方式中,可将气体组分送入蒸气回收系统1-7。该蒸气回收系统可用于例如回收流2-14内的任何期望的蒸气并经流2-15输送它们。另外,流2-16可用于输送来自蒸气回收系统的CO、CO2和/或其他不可回收的气体。应指出,在一些实施方式中,可在其他位置设置任选的蒸气回收系统。例如,在一些实施方式中,蒸气回收系统可位于吹扫流2-13的下游。本领域技术人员可选择蒸气回收系统的适当位置。可将其他产物(例如额外的气体)经流2-17输送到任选的压缩机1-9,在此可将它们压缩并用作反应器中的流化气体和/或它们可在此辅助将有机材料输送至反应器。在一些情况下,可进一步加工液体组分,例如将水相与有机相分离,以分离各化合物等。应理解,尽管图7中所述的该组实施方式包括反应器、固体分离器、再生器、冷凝器等,但并非所有实施方式都包括使用这些元件。例如,在一些实施方式中,进料流可被送入催化固体床反应器并反应,并可直接从反应器收集反应产物并冷却而不使用专用冷凝器。在一些情况下,尽管干燥器、研磨系统、固体分离器、再生器、冷凝器和/或压缩机可用作工艺的一部分,但这些元件中的一个或多个可包括不与反应器流体连接和/或一体连接的独立单元。在另一些实施方式中,干燥器、研磨系统、固体分离器、再生器、冷凝器和/或压缩机中的一个或多个可不存在。在一些实施方式中,可在制备工艺中的任何位置(例如,在通过反应器后、在分离后、在冷凝后等)回收所需的反应产物(例如,含氮芳香性化合物、烯烃、气体产物等)。在一些实施方式中,本发明的方法可包括使用多于一个反应器。例如,多个反应器可彼此流体连通,在一些实施方式中,该方法可包括在第一反应器中提供有机材料并在第一反应器中在足以产生一种或多种热解产物的反应条件下热解至少一部分该有机材料。在一些实施方式中,可向该第一反应器提供催化剂,并且在第一反应器中使用该催化剂在足以产生一种或多种流体产物的反应条件下使至少一部分所述一种或多种热解产物进行催化反应。该方法还可包括在第二反应器中使用催化剂在足以产生一种或多种流体产物的反应条件下使至少一部分所述一种或多种热解产物进行催化反应。在一些情况下,在第二反应器中使至少一部分所述一种或多种热解产物进行催化反应后,该方法可包括在第二反应器中使来自第一反应器的至少一部分一种或多种流体产物进一步反应以制备一种或多种其他产物的步骤。多反应器构造中的一个或多个反应器可包括流化床反应器(例如,循环流化床反应器、湍动流化床反应器等)和固定床反应器(如轴向绝热式固定床反应器、径向绝热式固定床反应器、列管式固定床反应器等),或在其他情况下任何其他类型的反应器(例如,上文提到的任何反应器)。例如,在一组实施方式中,第一反应器包括循环流化床反应器或湍动流化床反应器,且第二反应器包括与第一反应器流体连通的循环流化床反应器或湍动流化床反应器。另外,多反应器构造可包括本文提到的任何附加加工步骤和/或设备(例如,固体分离器、再生器、冷凝器等)。可利用本文提到的任何加工参数(例如,温度、停留时间等)操作反应器和/或附加加工设备。本发明中可用的有机材料可包括例如木糖醇、木糖、木聚糖、葡萄糖、纤维二糖、纤维素、淀粉、半纤维素、壳聚糖、甲壳素、蔗糖、果糖、木材、甘蔗渣、毛竹、玉米秸杆、废弃纸张、油菜籽饼、小桐子饼、饼粕、酒糟、废弃蛋白、微藻之类的组分,及其热解产物和此类组分和/或它们的热解产物的组合。有机材料的其他实例尤其包括,例如塑料废料、再生塑料、农业和城市固体废物、食品废物、动物废物、碳水化合物、木质纤维材料(例如,木屑或刨花、木质纤维生物质等)或它们的组合。有机材料还包括液体材料,如葡糖糖水溶液、甲基呋喃、2,5-二甲基呋喃、糠醛、5-羟甲基糠醛、5-甲基糠醛、γ-戊内酯、纤维素生物油、水溶性生物油、非水溶性生物油等,或它们的组合。生物油(一般生物油)主要来自于热解生物质原料产生的含氧碳氢化合物的浓缩液。将生物质在无氧或者缺氧条件下在适合温度(450-650℃)进行热解产生热解气,然后通过冷凝得到生物油。生物油是一种复杂的混合物,其中含有水,呋喃类化合物,乙酸、甲酸等有机酸,愈创木酚、丁香酚、苯酚、领苯二酚、香草醛等芳香类化合物。生物质主要包括上述原料。水溶性生物油主要来自于生物油水分离产物,将生物油与水按一定比率配制成混合物,然后将混合物在离心机中以一定转速离心一段时间至分层,将上层倒出就是水溶性生物油,剩余部分是水不溶性生物油。如本文所示,有机材料和催化剂材料的选择可用于改变所得流体产物的组成。如上所述,进料组合物中有机材料可包括固体、液体和/或气体。在有机材料包括固体的情况下,固体可具有任何合适的尺寸。在一些情况下,使用具有较小粒度的有机材料固体可能是有利的。由于它们与较大固体相比具有较高的表面积/体积比,小颗粒固体在一些情况下可比较大固体反应快。另外,小粒度可实现在各颗粒内和/或反应器容积内更高效的热传递。这可防止或降低不想要的反应产物的形成。另外,小粒度可提高固体-气体和固体-固体接触,从而改进热和质量的传递。在一些实施方式中,固体有机材料的平均尺寸小于约5毫米,小于约2毫米,小于约1毫米,小于约500微米,小于约60目(250微米),小于约100目(149微米),小于约140目(105微米),小于约170目(88微米),小于约200目(74微米),小于约270目(53微米),或小于约400目(37微米)或更小。在一些情况下,希望使用平均粒度高于最小量的进料材料以降低使有机材料进料通过反应器所需的压力。例如,在一些情况下,希望使用平均粒度为至少约400目(37微米),至少约270目(53微米),至少约200目(74微米),至少约170目(88微米),至少约140目(105微米),至少约100目(149微米),至少约60目(250微米),至少约500微米,至少约1毫米,至少约2毫米,至少约5毫米或更高的固体有机材料。可从本领域中已知的任何催化剂中选择可用于本发明中的催化剂组分,或如了解本发明的本领域技术人员理解的那样选择可用于本发明的催化剂组分。在功能上,催化剂可能只受限于任何此类材料的能力,促进和/或影响脱水、脱氢、氨化、异构化、氢转移、芳构化、脱羰作用、脱羧、醇醛缩合和/或其他任何与有机材料热解过程相关的反应。如本领域技术人员理解的那样,认为催化剂组分可为酸性、中性或碱性。在一些情况下,本文所述的催化剂颗粒可包括多晶固体(例如,多晶颗粒)。在一些实施方式中,催化剂颗粒还可包括单晶。在某些情况下,所述颗粒可为不同的可单独的和分离的物理物体。在其他情况下,所述颗粒可为至少在其制备和/或使用中的多个单独的颗粒的某些点相互紧密接触形成的团块。用于本文所述的实施方式中(例如,在进料流中,在反应器中等)的催化剂可具有任何合适的尺寸。在一些情况下,使用较小颗粒的催化剂是有利的,如先前所提到的,在一些实施方式中,所述催化剂颗粒可为包括多个成团的较大的催化剂颗粒物体形式。在一些实施方式中,例如,使用小催化剂颗粒可增加有机材料与催化剂表面位点接触的程度,原因是外部催化表面积增加以及通过催化剂的扩散距离减小。在一些情况下,可至少部分基于例如所需的流体流的类型和催化剂的寿命来选择催化剂尺寸和/或催化剂粒度。在一些实施方式中,在某些情况下包括单个的催化剂颗粒或在其他情况下包括多个颗粒的团块的催化剂物体的平均直径(通过常规的筛析所检测)可为小于约5毫米,小于约2毫米,小于约1毫米,小于约500微米,小于约60目(250微米),小于约100目(149微米),小于约140目(105微米),小于约170目(88微米),小于约200目(74微米),小于约270(53微米),或小于约400目(37微米)或更小。在一些实施方式中,催化剂物体可为具有以下最大横截面尺寸的颗粒或由其形成:小于约5微米、小于约1微米、小于约500nm、小于约100nm、约100nm至约5微米、约500nm至约5微米、约100nm至约1微米、或约500nm至约1微米。如先前所述,在某些情况下,具有前文所述范围的尺寸的催化剂颗粒可成团而形成可分离的具有上一段所述范围的尺寸的催化剂物体。本文所用的颗粒的“最大横截面尺寸”是指颗粒的两个边界之间的最大尺寸。本领域普通技术人员能够通过例如分析催化剂制剂的扫描电子显微图像(SEM)来测量颗粒的最大横截面尺寸。在成团颗粒的实施方式中,当测定最大横截面尺寸时应分开考虑颗粒。在这样的情况下,通过建立各成团颗粒之间的虚构边界并测量由这样的边界而产生的假定的个体化颗粒的最大横截面尺寸来进行测量。在一些实施方式中,催化剂内较大量的颗粒的最大横截面尺寸位于给定的范围内。例如,在一些实施方式中,催化剂内至少约50%、至少约75%、至少约90%、至少约95%、或至少约99%的颗粒的最大横截面尺寸为小于约5微米、小于约1微米、小于约500nm、小于约100nm、约100nm至约5微米、约500nm至约5微米、约100nm至约1微米、或约500nm至约1微米。在一些情况下,在特定范围内具有最大横截面尺寸的颗粒可占催化剂中较大比例的体积比。例如,在一些实施方式中,具有如下最大横截面尺寸的颗粒:小于约5微米、小于约1微米、小于约500nm、小于约100nm、约100nm至约5微米、约500nm至约5微米、约100nm至约1微米、或约500nm至约1微米占所使用的所有催化剂的体积总和的至少约50%、至少约75%、至少约90%、至少约95%或至少约99%。在一些实施方式中,催化剂内的颗粒基本上具有相同尺寸。例如,催化剂可包括具有如下尺寸分布的颗粒:该尺寸分布使得颗粒的最大横截面尺寸的标准差不大于颗粒的平均最大横截面尺寸的约50%、不大于约25%、不大于约10%、不大于约5%、不大于约2%或不大于约1%。标准差(小写的σ)是其在本领域中的常规含义,可如下计算:其中,Di是颗粒i的最大横截面尺寸,Davg是所有颗粒的平均最大横截面尺寸,n是催化剂内的颗粒数量。上述标准差与颗粒的平均最大横截面尺寸之间的百分比比较可通过标准差除以平均值并乘以100%获得。使用包括位于上述所选尺寸分布的颗粒的催化剂可导致由有机材料反应所产生的含氮芳香性化合物的产率和/或选择性增加。例如,在一些情况下,相对于使用含有处于所需范围之外的尺寸分布的颗粒的催化剂所产生的含氮芳香性化合物的量(例如,使用大比例的大于1微米、大于5微米等的颗粒),使用含有具有所需尺寸范围(例如,上述任何尺寸分布)的颗粒的催化剂可导致反应产物中含氮芳香性化合物的量增加至少约5%,至少约10%,或至少约20%。可选地,单独或与上述考虑相结合,根据孔径可以选择催化剂(例如,通常与沸石相关的介孔型和孔径),例如小于约100埃、小于约50埃、小于约20埃、小于约10埃、小于约5埃或更小的平均孔径。在一些实施方式中,可使用平均孔径为约5埃至约100埃的催化剂。在一些实施方式中,可使用平均孔径为约5.5埃至约6.5埃,或约5.9埃至约6.3埃的催化剂。在一些情况下,可使用平均孔径为约7埃至约8埃或约7.2埃至约7.8埃的催化剂。本文所用的术语“孔径”用于表示孔的最小横截面直径。孔的最小横截面直径可对应于与孔长度垂直地测得的最小横截面尺寸(例如,横截面直径)。在一些实施方式中,“平均孔径”或“孔径分布”为X的催化剂是指催化剂内孔的最小横截面直径的平均值为约X的催化剂。应理解,本文所用的孔的“孔径”或“最小横截面直径”是指本领域技术人员公知的Norman半径调节孔径。本领域普通技术人员会理解如何测定催化剂中的孔径(例如,最小孔径、最小孔径的平均值)。例如,XRD可用于测定孔径(例如,沸石孔径)的其他技术包括,例如氦测比重术或低压氩吸附技术。除非另有指明,否则本文中提到的孔径为通过x-射线衍射测定的那些,其如上所述校正以反映它们的Norman半径调节孔径。在一些实施方式中,应用筛选法选择具有适于转化特定热解产物分子的孔径的催化剂。筛选法可包括确定进行催化反应的热解产物分子的尺寸(例如,热解产物分子的分子动力学直径)。本领域普通技术人员可计算例如给定分子的动力学直径。催化剂的类型则可如下进行选择,使得催化剂的孔隙(例如Norman调节最小半径)大到足以使热解产物分子扩散至催化剂中和/或与催化剂反应。在一些实施方式中,可选择催化剂使得它们的孔径小至足以防止其他不希望反应的热解产物进入和/或反应。非限制性地,一些这样的和其他催化剂可选自天然存在的沸石、合成沸石及它们的组合。在一些实施方式中,如本领域技术人员理解的那样,催化剂可为丝光沸石结构转化(MordeniteFrameworkInverted,MFI)型沸石催化剂,例如ZSM-5沸石催化剂。任选地,此类催化剂可包括酸性位点。其他类型的沸石催化剂尤其包括镁碱沸石、沸石Y、沸石β、丝光沸石、MCM-22、ZSM-23、ZSM-57、SUZ-4、EU-1、ZSM-11、(S)AlPO-31、SSZ-23。在其他实施方式中,可使用非沸石催化剂,例如,WOx/ZrO2、硫酸铝等。在一些实施方式中,催化剂可包括金属和/或金属氧化物。合适的金属和/或氧化物尤其包括,例如镍、铂、钒、钯、锰、钴、锌、铜、铬、镓和/或它们的任何氧化物。在一些实施方式中,可将金属和/或金属氧化物浸入催化剂(例如,在催化剂的晶格结构的空隙中)。可将金属和/或金属氧化物掺入催化剂的晶格结构中。例如,在催化剂的制备过程中,可包括金属和/或金属氧化物,且金属和/或金属氧化物可占据所产生的催化剂(例如,沸石催化剂)的晶格部位。作为另一个实例,金属和/或金属氧化物可与沸石反应或否则与其相互作用,使得金属和/或金属氧化物取代沸石的晶格结构内的原子。在某些实施方式中,可使用包括镓的丝光沸石结构转化(MFI)沸石催化剂。例如,可使用镓铝硅酸盐MFI(GaAlMFI)沸石催化剂。本领域普通技术人员熟悉可被认为是铝硅酸盐MFI沸石的GaAlMFI沸石,其中,一些Al原子被Ga原子取代。在一些情况下,沸石催化剂可为氢形式(例如,H-GaAlMFI)。在一些实施方式中,镓铝硅酸盐MFI催化剂可为其中一些铝原子被镓原子取代的ZSM-5沸石催化剂。在一些情况下,镓铝硅酸盐沸石催化剂中的Si摩尔数与镓铝硅酸盐沸石催化剂中的Ga和Al的总摩尔数之比(即以Si:(Ga+Al)表示的摩尔比)可为至少约15:1、至少约20:1、至少约25:1、至少约35:1、至少约50:1、至少约75:1或更高。在一些实施方式中,使用沸石中Si的摩尔数与Ga和Al的总摩尔数之比如下的催化剂是有利的:约15:1至约100:1、约15:1至约75:1、约25:1至约80:1或约50:1至约75:1。在一些情况下,镓铝硅酸盐沸石催化剂中的Si与镓铝硅酸盐沸石催化剂中的Ga的摩尔比可为至少约15:1、至少约60:1、至少约120:1、至少约200:1、约30:1至约300:1、约15:1至约200:1、约15:1至约120:1、或约15:1至约75:1。镓铝硅酸盐沸石催化剂中的Si与镓铝硅酸盐沸石催化剂中的Al的摩尔比可为至少约10:1、至少约20:1、至少约30:1、至少约40:1、至少约50:1、至少约75:1、约10:1至约100:1、约10:1至约75:1、约10:1至约50:1、约10:1至约40:1、或约10:1至约30:1。另外,在一些情况下,可选择催化剂的性质(例如,孔隙结构、酸性位点的类型和/或数量)以选择性地制备所需产物。在一些实施方式中,可能希望使用一种或多种催化剂以建立双峰孔径分布。在一些情况下,可使用具有具有双峰孔径分布的单个催化剂(例如,主要含有5.9-6.3埃孔隙和7-8埃孔隙的单个催化剂)。在其他情况下,可使用两种或多种催化剂的混合物以建立双峰分布(例如,两种催化剂的混合物,各催化剂类型包括不同范围的平均孔径)。在一些实施方式中,一种或多种催化剂的一种可包括沸石催化剂,且一种或多种催化剂的另一种包括非沸石催化剂(例如,介孔型催化剂、金属氧化物催化剂等)。例如,在一些实施方式中,一种或多种催化剂(例如,沸石催化剂、介孔型催化剂等)的至少约70%、至少约80%、至少约90%、至少约95%、至少约98%或至少约99%的孔隙具有在第一尺寸分布或第二尺寸分布内的最小横截面直径。在一些情况下,一种或多种催化剂的至少约2%、至少约5%或至少约10%的孔隙具有在第一尺寸分布内的最小横截面直径;且一种或多种催化剂的至少约2%、至少约5%或至少约10%的孔隙具有在第二尺寸分布内的最小横截面直径。在一些情况下,第一尺寸分布和第二尺寸分布选自上文提供的范围。在一些实施方式中,第一尺寸分布和第二尺寸分布彼此不同并且不重叠。非重叠范围的一个实例是5.9-6.3埃和6.9-8.0埃,重叠范围的一个实例是5.9-6.3埃和6.1-6.5埃。可选择第一尺寸分布和第二尺寸分布以使范围不彼此紧邻,例如5.9-6.3埃和6.9-8.0埃的孔径。范围彼此紧邻的一个实例是5.9-6.3埃和6.3-6.7埃的孔径。作为一个具体实例,在一些实施方式中,使用一种或多种催化剂提供双峰孔径分布以同时制备含氮芳香性化合物和烯烃化合物。也就是说,一种孔径分布可有利地制备较高量的含氮芳香性化合物,另一孔径分布可有利地制备较高量的烯烃化合物。在一些实施方式中,所述一种或多种催化剂的至少约70%、至少约80%、至少约90%、至少约95%、至少约98%或至少约99%的孔隙具有约5.9埃至约6.3埃或约7埃至约8埃的最小横截面直径。另外,所述一种或多种催化剂的至少约2%、至少约5%或至少约10%的孔隙具有约5.9至约6.3埃的最小横截面直径;且所述一种或多种催化剂的至少约2%、至少约5%或至少约10%的孔隙具有约7埃至约8埃的最小横截面直径。在一些实施方式中,所述一种或多种催化剂的至少约70%、至少约80%、至少约90%、至少约95%、至少约98%或至少约99%的孔隙具有约5.9埃至约6.3埃,或约7埃至约200埃的最小横截面直径。另外,所述一种或多种催化剂的至少约2%、至少约5%或至少约10%的孔隙具有约5.9埃至约6.3埃的最小横截面直径;且所述一种或多种催化剂的至少约2%、至少约5%或至少约10%的孔隙具有约7埃至约200埃的最小横截面直径。在一些实施方式中,所述一种或多种催化剂的至少约70%、至少约80%、至少约90%、至少约95%、至少约98%或至少约99%的孔隙具有在第一分布和第二分布内的最小横截面直径,其中第一分布为约5.9埃至约6.3埃,第二分布与第一分布不同且不重叠。在一些实施方式中,第二孔径分布可为约7埃至约200埃,约7埃至约100埃,约7埃至约50埃,或约100埃至约200埃。在一些实施方式中,第二催化剂可为介孔型的(例如,具有约2纳米至约50纳米的孔径分布)。在一些实施方式中,双峰孔径分布可有益于使两种或多种有机材料组分进料反应。例如,一些实施方式包括在反应器中提供包括第一组分和第二组分的固体有机材料,其中第一组分和第二组分不同。可用作第一组分或第二组分的化合物的实例包括本文所述的任何有机材料(例如,甘蔗渣、葡萄糖、木材、毛竹、玉米秸杆、纤维素、半纤维素、木质素或任何其他有机材料)。例如,所述第一组分可包括纤维素、半纤维素和木质素之一,且所述第二组分包括纤维素、半纤维素和木质素之一。该方法还包括在反应器中提供第一催化剂和第二催化剂。在一些实施方式中,所述第一催化剂可具有第一孔径分布,且所述第二催化剂可具有第二孔径分布,其中第一孔径分布和第二孔径分布不同且不重叠。第一孔径分布可为例如约5.9埃至约6.3埃。第二孔径分布可为例如约7埃至约200埃,约7埃至约100埃,约7埃至约50埃,或约100埃至约200埃。在一些情况下,所述第二催化剂可为介孔型的或无孔的。第一催化剂可选择性地使第一组分或其衍生物进行催化反应以制备流体产物。另外,第二催化剂可选择性地使第二组分或其衍生物进行催化反应以制备流体含氮芳香性产物。该方法还可包括在足以产生一种或多种热解产物的反应条件下在反应器中热解至少一部含有机材料,和用第一催化剂和第二催化剂使至少一部分热解产物进行催化反应以制备一种或多种含氮芳香产物。在一些情况下,也可产生至少部分失活的催化剂。在一些实施方式中,与本文所述的实施方式联用的方法包括提高组合物的催化剂与有机材料质量比,以提高可识别的含氮芳香性化合物的生成。如本文所示(代表但有别于某些现有催化热解法),本文所述的制品和方法可用于制备分离的、可识别的含氮芳香性化合物,选自但不限于吡嗪、甲基吡嗪、吡啶、甲基吡啶、苯胺、甲基苯胺、吲哚、甲基吲哚及它们的组合。在一些实施方式中,可通过添加一种或多种另外的化合物来影响催化剂的反应化学。例如,将金属添加至催化剂中可导致特定化合物的选择性形成的转变(例如,将金属添加到氧化铝-硅酸盐催化剂中可导致生成更多CO)。另外,当流化流体包含氢时,可降低催化剂上形成的焦炭量。在一些实施方式中,催化剂可包括二氧化硅和氧化铝(例如,沸石催化剂)。催化剂中的二氧化硅和氧化铝可以以任何合适的摩尔比存在。在一些实施方式中,应用相对于氧化铝的摩尔数具有更大摩尔数的二氧化硅(即高的二氧化硅与氧化铝摩尔比)的催化剂是有利的。本发明人意外地发现,例如与本文所述的实施方式结合,高的二氧化硅与氧化铝摩尔比可导致形成较大量的含氮芳香性化合物产物。例如,在一些情况下,进料组合物可包括至少约15:1、至少约30:1、至少约40:1、至少约50:1、至少约75:1、至少约100:1、至少约150:1或更高的二氧化硅与氧化铝摩尔比。在一些实施方式中,使用二氧化硅与氧化铝摩尔比为约15:1至约200:1、约30:1至约150:1、约50:1至约160:1、或约100:1至约150:1的催化剂是有利的。在一些实施方式中,催化剂和有机材料可以以任何合适的比率存在。例如,在进料组合物(例如,通过包括催化剂和有机材料的一个或多个进料流或通过分开的催化剂和有机材料进料流)包括催化剂和有机材料(例如,循环流化床反应器)的情况下,催化剂和有机材料可以以任何合适的质量比存在。作为另一实例,在一开始在反应器中装载催化剂和有机材料的混合物(例如,间歇式反应器)的情况下,催化剂和有机材料可以以任何合适的质量比存在。在涉及循环流化床反应器的一些实施方式中,进料流中(即向反应器提供的包括固体催化剂和固体有机材料的组合物中)催化剂与有机材料的质最比可为至少约0.5:1、至少约1∶1、至少约2:1、至少约5:1、至少约10:1、至少约15:1、至少约20:1或更高。在涉及循环流化床反应器的一些实施方式中,进料流中催化剂与有机材料的质量比可为小于约0.5:1、小于约1:1、小于约2:1、小于约5:1、小于约10:1、小于约15:1或小于约20:1;或约0.5:1至约20:1、约1:1至约20:1、或约5:1至约20:1。应用较高的催化剂与有机材料质量比可有利于由进料材料热解形成的挥发性有机化合物在热分解成焦炭之前引入催化剂。当催化剂与有机材料的质量比小于约5:1时,主要产物为吡咯类化合物和吡嗪类化合物;当催化剂用量提高,吡咯类以及吡嗪类化合物选择性下降,吲哚类化合物,苯胺类化合物以及吡啶类化合物提高。当催化剂与有机材料比例约10:1时,产物主要为吲哚类化合物和苯胺类化合物,提高二氧化硅与氧化铝摩尔比,吡啶类化合物选择性提高。不受任何理论限制,这种效应可至少部分是由于反应器中存在化学计量过量的催化剂位点而导致的。在一些实施方式中,如反应器为固定床反应器时,空速(液体进料与催化剂的质量归一化比例)可以以任何合适的比率存在。例如进料为液体原料时,相对于单位催化剂体积的液体进料量(WHSV),以0.05至10的WHSV,如小于约0.05、小于约0.1、小于约0.2、小于约0.5、小于约0.8、小于约1、小于约1.5、小于约2、小于约5、小于约10进料至反应器。当WHSV大于约为2时,主要产物为吡咯类化合物,吡啶类化合物;随着WHSV下降,吡咯类化合物选择性降低,吲哚类化合物和苯胺类化合物选择性提高。在一些实施方式中,本文所述的制品和方法构造成在例如在单级热解设备中或另外可选的多级热解设备中选择性地制备含氮芳香性化合物。流体产物可包括例如含氮芳香性化合物的量占固体有机材料的总反应产物的至少约10wt%,至少约15wt%,至少约20wt%,至少约25wt%,至少约30wt%,至少约35wt%,至少约39wt%,约10wt%至约40wt%,约10wt%至约35wt%,约15wt%至约40wt%,约15wt%至约35wt%,约20wt%至约40wt%,约20wt%至约35wt%,约25wt%至约40wt%,约25wt%至约35wt%,约30wt%至约40wt%,或约30wt%至约35wt%。上述化合物产率以碳产率计即通过将产物中的碳摩尔数除以进料中的碳摩尔数来计算碳产率。以给定产物中的碳摩尔数除以所有产物(不包括CO、CO2和焦炭(例如,留在催化剂上的固体焦炭))中的碳摩尔数来计算选择性。应指出,本领域普通技术人员能够在重量百分比和碳产率之间进行转换。例如,可通过化学分析测定碳质材料进料中的碳量。此外,可利用它们的分子式计算各反应产物的碳百分比。本文所用的术语“含氮芳香性化合物”用于表示包括一个或多个具有芳香性环基,例如单芳环体系(例如吡嗪、吡啶、吡咯等)和稠多环芳环体系(例如吲哚等)的烃化合物。含氮芳香性化合物的实例包括但不限于,吡嗪、2-甲基吡嗪、2,5-二甲基吡嗪、2,6-二甲基吡嗪、吡啶、2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、、二甲基吡啶、吡咯、2-甲基吡咯、3-甲基吡咯、2,5-二甲基吡咯、吲哚、1-甲基吲哚、2-甲基吲哚、3-甲基吲哚、2,8-二甲基吲哚、苯胺、邻甲基苯胺、间甲基苯胺、对甲基苯胺、二甲基苯胺等或它们组合中一种或几种。在一些实施方式中,可制备单环和/或更多环的含氮芳香性化合物。含氮芳香性化合物可具有例如C5-C14、C6-C8、C6-C12、C8-C12、C10-C14的碳数,N1-N2、N3-N4、N5-N6的氮数。吡嗪类化合物可以作为重要的医药中间体和香精香料中间体,也是一种具有高附加值的精细化学品。吡啶及其衍生物是非常重要的化工中间体,广泛应用于医药、农药、饲料、合成橡胶和印染工业,还可用于表面活性剂和食品添加剂的生产。吡咯类化合物作为精细化工产品的重要中间体在医药、食品、农药、日用化学品、涂料、纺织、印染、造纸、感光材料高分子材料等领域具有广泛用途。吲哚是基本化工原料,广泛用于医药、农药、染料、食品以及香料等众多领域。苯胺是一种重要的工业化学品,可作为橡胶硫化促进剂、染料、媒染体、药品、炸药原料,和二苯基甲烷聚氨酯(MDI)。在本发明的一些实施方案中,可以将以上含氮芳香性化合通过本领域公知的方法分离,例如可以采用蒸馏(包括常压蒸馏、减压蒸馏、分子蒸馏)、精馏、柱层析色谱法、酸抽提法、离子变换色谱法、萃取法等方法。在本发明中,根据不同的催化方法可以有选择性地制备不同化合物。首先可以根据化合物的本身物性参数(见表一)进行蒸馏,可以得到不同温度的馏分。如吡嗪类馏分,吡咯类馏分,吡啶类馏分、苯胺类馏分和吲哚类馏分。然后再进行精馏分离产物得到更高纯度的产品。有些化合物之前沸点相差很低,如2-甲基吲哚与3-甲基吲哚,可以根据其极性大小,采用柱层析色谱法等进行分离。有些化合物之间沸点相近,但是分子量相差很大,可以采用分离蒸馏方法。另外,在一些实施方式中,可选择促进含氮芳香性化合物的选择性制备的催化剂。例如,在一些情况下,ZSM-5可优先制备较高量的含氮芳香性化合物。在一些情况下,包括Bronstead酸位点的催化剂可促进含氮芳香性化合物的选择性制备。另外,具有良序孔隙结构的催化剂可促进含氮芳香性化合物的选择性制备。例如,在一些实施方式中,平均孔径为约5.9埃至约6.3埃的催化剂可特别用于制备含氮芳香性化合物。另外,平均孔径为约7埃至约8埃的催化剂可用于制备烯烃。在一些实施方式中,使用一个或多个上述工艺参数的组合以促进含氮芳香性化合物的选择性制备。制成的含氮芳香性化合物与烯烃的比例可为例如约0.1:1至约10:1、约0.2:1至约5:1、约0.5:1至约2:1、约0.1:1至约0.5:1、约0.5:1至约1:1、约1:1至约5:1、或约5:1至约10:1。实施例在以下实施例中,对于反应产物的表征主要采用气相色谱法,气相色谱-质谱法,其中,主要使用杭州科晓仪器制造的GC-1690气相色谱仪,采用30m×0.25mm×0.25μm色谱柱,以10℃/min程序升温至280℃。气相-质谱联用仪主要使用美国ThermoTraceGCUltrawithanISQimassspectrometer,采用TR-35MS色谱柱(30m×0.25mm×25um),检测方法是在40℃保持3min,然后以5℃/min升温至180℃,再以10℃/min升温至280℃。下列非限制性实施例和数据旨在说明与本发明的方法和/或组合物相关的各个方面和特征,包括通过本文所述的热解法可获得的各种化合物的选择性制备,但并非列举本发明的整个范围。与现代技术相比,本发明的方法和组合物提供令人惊讶的、意外的结构和数据。尽管通过使用几种催化剂和原料例证本发明的效用,但本领域技术人员会理解的是,用与本发明的范围相当的各种其他催化剂材料和/或原料可获得相当的结果。实施例1在此实施例中使用直径34mm,长度300mm的石英管式反应器。在反应器中,催化剂由石英棉支承。将石英反应器装在温控炉中。在操作过程中,使用氨气作为载气,通过气体流量计控制其流速。本实验载气采用NH3或者NH3/N2、NH3/He混合气。粉状原料与载气流一起从石英管开口处流至热解界面。液体产物从反应器流至冷凝器,气体产物收集在气体采样袋中。使用气相色谱仪分析液体和气体产物。(模型反应图见图5)作为若干实施方式的代表,用粉状催化剂和进料(<140筛目)进行实施例1-10中所述的催化热解试验。除非此实施例中另行说明,否则这些反应条件由上所述。通过物理混合碳水化合物进料和催化剂制得粉状反应物。以葡萄糖与HZSM-5(Si/Al=25),制备催化剂与进料质量比为2的葡萄糖的物理混合物。还制备催化剂与进料质量比为2的纤维二糖/HZSM-5,纤维素/HZSM-5,木糖/HZSM-5,木糖醇/HZSM-5,木聚糖/HZSM-5,壳聚糖/HZSM-5,甲壳素/HZSM-5,蔗糖/HZSM-5,果糖/HZSM-5,产物分布GC/MS图见附图1。实施例2将催化剂添加到热解工艺中显著降低焦炭形成并提高相热稳定产物的转化率。测试6种不同催化剂用于葡萄糖的催化热解,包括:γ-氧化铝(γ-Al2O3),二氧化硅氧化铝(SiO2-Al2O3),氧化钨氧化锆(WO3/ZrO2),硫酸二氧化锆(SO42-/ZrO2),MCM-41,HZSM-5(Si/Al=63)。通过物理混合碳水化合物进料和催化剂制得粉状反应物,本实验催化剂与葡萄糖的用量比为2,反应温度为550℃,气体流速为200mi/min.催化剂与葡萄糖混合后研磨成粉末。催化剂结构极大地改变产物选择性,利用HZSM-5催化剂可提高吡咯类化合物选择性,利用SO42-/ZrO2可以提高吡嗪类化合物选择性。实施例3该实施例研究了反应温度对产物产率的影响。表4和表5表明随着温度的升高对氮杂环产物有所提高,但是随着温度升高吡咯类化合物的选择性有所下降,吲哚类化合物和苯胺类化合物的选择性会有所提高。实施例4该实施例研究了改变催化剂的二氧化硅与氧化铝摩尔比的影响。这些实验的条件如下:催化剂/进料质量比为5;催化剂,HZSM-5;反应温度550℃;载气速率200ml/min。使用葡萄糖作为这些实验的原料。表6显示不同二氧化硅与氧化铝摩尔比对葡萄糖催化热解的产物选择性。如表6所示低硅铝比可以提高吡咯的选择性,硅铝比提高有利于提高吡啶类化合物的选择性。实施例5该实施例研究了改变催化剂与葡萄糖的进料量比对产物收率以及产物分布的影响。这些实验的条件如下:催化剂,HZSM-5;反应温度500℃;载气速率150ml/min。使用葡萄糖作为这些实验的原料。表7显示作为二氧化硅与氧化铝摩尔比的函数的葡萄糖催化热解的产物选择性。如表7所示,催化剂与进料量比分别为1、2、5、10、15、20。由表7和8表明,随着催化剂用量的增加,吡咯类产物和吡嗪类产物逐渐降低,吲哚类产物逐渐增加。实施例6实施例研究了改变载气中NH3与N2比率对产物收率以及产物分布的影响。这些实验的条件如下:催化剂,HZSM-5;反应温度500℃;以NH3与N2混合气为载气,载气速率250ml/min。使用葡萄糖作为这些实验的原料。本实验载气NH3/N2为0:1,1:19,1:9,1:4,1:1,4:1,19:1以及1:0.表9显示NH3与N2分布对的葡萄糖催化热解的产物选择性。由表9表明,产物中含氮化合物与载气中NH3有着很重要关系,随着NH3量增加,相应含氮化合物的量也增加。实施例7催化剂中的金属浸渍对产物产率存在影响。用金属浸渍HZSM-5(二氧化硅与氧化铝摩尔比为25)孔隙使产物选择性移向CO和CO2,表明金属可影响化学反应。不希望受到任何理论限制,金属可提高脱碳和/或脱羧反应速率。对下列金属进行测试。除了以下所述金属,本专利还包含其他金属如:In,Ru,Rh,Ir,Pt,Pd,Au,Re,Tl,La系。表10概述了掺杂金属的HZSM-5上的葡萄糖催化热解获得的结果。利用两种不同方法将金属掺杂至HZSM-5中:干/湿法浸渍和离子交换。使用离子交换法浸渍的催化剂比使用干/湿法浸渍法浸渍的催化剂产生更高的吡咯类化合物和吲哚类化合物产率。实施例8催化剂孔径同样影响含氮杂环化合物的收率。使用葡萄糖作为这些实验的原料。表11表示在几种不同的沸石结构上催化热解葡萄糖的碳产率数据。这些实验的条件如下:催化剂,反应温度500℃;载气速率200ml/min,催化剂与葡萄糖进料比为5。从表11中的结果可以看出,孔径接近ZSM-5有利于产生含氮杂环化合物。实施例9天然形成的生物质也可用于实验中的原料通过催化热解制备吡咯类化合物和吲哚类化合物。表12概述天然形成的生物质催化热解的结果。木材、甘蔗渣、玉米秸秆、废弃纸张、油菜籽饼、微藻使用HZSM-5催化剂热解产生含氮化合物、不含氮化合物、产生气体以及焦炭。这些结果表明,可以用天然形成的生物质催化热解制备吡咯类化合物和吲哚类化合物等含氮化合物。实施例10通过物理混合碳水化合物进料和催化剂制得粉状反应物。制备催化剂与进料质量比为10的HZSM-5/葡萄糖,HZSM-5/纤维素以及HZSM-5/木糖,木糖醇/HZSM-5,木聚糖/HZSM-5,果糖/HZSM-5,蔗糖/HZSM-5,壳聚糖/HZSM-5,甲壳素/HZSM-5混合物制备吲哚类产物和苯胺类化合物。这些实验的条件如下:催化剂为HZSM-5;催化剂与进料量比为10,进料速度为0.2g/min。产物分布GC/MS图见附图2.实施例11上述实施例主要采用的试验方法为流化床方式,催化剂与原来混合研磨后,制备成140筛目的粉末进样;本实验还可以采用固定床方式进样,试验方式如下:在此实施例中,使用直径34mm,长度300mm的石英管式反应器,在反应器中填充HZSM-5催化剂以产生固定床,催化剂两端由石英棉支承,将石英反应器装在温控炉中。在操作过程中,使用氨气作为载气,通过气体流量计控制其流速。粉状原料与氨气载气流一起从石英管开口处流至热解界面。液体产物从反应器流至的冷凝器,气体产物收集在气体采样袋中。使用气相色谱仪分析液体和气体产物。本实施例主要采用固定床分别制备吡嗪类化合物、吡咯类化合物、吲哚类化合物以及苯胺类化合物。实验方法如下,分别制备催化剂质量分别为2g,5g和10g固定床反应器,将反应至于反应器中,反应原料随载气一同进入催化剂床层,液体产物流至冷凝器,气体产物收集在气体采样袋中。使用气相色谱仪分析液体和气体产物。表14明制备含氮化合物,表15明液体中各种化合物的碳选择性。实施例13上述实施例主要描述固体进样方式,包括催化剂和反应原料混合后流动床反应方式和固定床反应方式;此实施例描述了固定床、流动反应器系统的使用。在此实施例中,使用直径为10mm,长度为250mm的石英管式反应器。在此反应器中装载一定量(0.01g-2.5g)HZSM-5催化剂以产生固定床,其由石英棉支撑,将石英反应器装在高温炉中。本实验载气采用NH3或者NH3/N2、NH3/He混合气。反应原料为液体原料,通过质量流量控制器控制其流速。通过注射泵将液体原料引入载气中进入反应器。液体产物从反应器流至冷凝器,气体产物收集在气体采样袋中。使用气相色谱仪分析液体和气体产物。本实施例中,以空速(WHSV)即液体进料质量(g/h)除以催化剂重量(g)计算质量归一化为空速。通过产物中的碳摩尔数除以输入反应器的碳摩尔数,计算碳产率。作为若干实施方式的代表,进行实施例14-16中所述的催化热解试验。除非实施例中另行说明,否则这些反应条件由上所述。实施例14本实施例主要研究液体反应、原料的影响。这些实验的条件如下:催化剂为ZSM-5,催化剂用量为1g,反应温度500℃;载气为NH3,载气速率40ml/min。本实验中纤维素生物油和生物油分别来自于500℃条件下热解纤维素、杨木、微藻等得到。本实施例以松木和纤维素为代表,其他生物质同样适用于本实验原料要求。水溶性生物油主要来自于生物油水分离产物,生物油与水比率为1∶4配制成混合物,然后将混合物在离心机中以1000转/分钟转速离心20分钟,将上层倒出就是水溶性生物油,剩余部分是水不溶性生物油。(反应流程模型图见图6)实施例15本实施例主要以呋喃为模型化合物研究WHSV对产物分布的影响。实验的条件如下:催化剂为HZSM-5(Si/Al=25),催化剂用量为1g,反应温度500℃;载气为NH3,载气速率20ml/min。由表17可知,随着WHSV变小,吡咯和甲基吡咯的选择性下降,吡啶类化合物,吲哚类化合物以及苯胺类化合物的选择性提高。由于生物油中主要化合物为呋喃基化合物,因此本实施例以呋喃为模型化合物。降低WHSV,使得反应更加充分产物向有利于吲哚方向产生,提高WHSV利于产生吡咯类化合物。实施例16本实施例主要以呋喃为模型化合物研究温度对产物分布的影响。实验的条件如下:催化剂为HZSM-5(Si/Al=25),催化剂用量为1g;载气为NH3,载气速率40ml/mm,WHSV是1h-1。本实施例的产物主要包括吡啶类化合物,吡咯类化合物,苯胺类化合物以及吲哚类化合物。由表18可知,温度升高,吡咯和甲基吡咯的选择性下降,吡啶类化合物,吲哚类化合物以及苯胺类化合物的选择性提高。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1