紫甘薯多糖的提取方法及其用途与流程
本发明涉及紫甘薯多糖的提取方法及其用途,具体地说,涉及紫甘薯多糖的提取方法以及该多糖在制备抗氧化的功能性食品中的用途。
背景技术:
紫甘薯(IpomoeabatatasPoir)又称川山紫,原产于美洲热带地区,属于甘薯的一种,旋花科甘薯属蔓生草本植物,生长环境范围广、容易栽培。紫甘薯富含各种微量元素、氨基酸、蛋白质、多糖等成分,具有抗突变和抗氧化,缓解肝功能障碍及抗肿瘤等保健功能,紫甘薯水提液具有一定的抗突变和抗氧化能力,其中多糖具有广泛的药理作用,如作为免疫调节剂,激活巨噬细胞,T淋巴细胞等多种免疫细胞,促进细胞因子生成,抗病毒、抗感染、抗氧化、抗衰老、抗肿瘤等,其中关于多糖抗肿瘤活性的研究主要是通过体内实验来证明的,对于其抗肿瘤的分子机制还未见报道。目前国内外已经进行了大量紫甘薯色素提取研究,但对紫甘薯多糖(polysaccharidefrompurplesweetpotato,PPSP)的提取研究未见报道。OPG/RANK/RANKL系统最初是在研究与骨相关的一些疾病如骨质疏松症和类风湿性关节炎中被发现的,而RANKL在破骨细胞形成和骨质吸收中起中心作用。最近有文献报道,该系统在导致乳腺癌转移的过程中起重要作用,而RANKL是促进乳腺癌转移的重要因素之一。RANKL的生物活性主要是以三聚体的状态存在,如果紫甘薯多糖与RANKL作用后改变了RANKL的聚集状态就会影响RANKL与RANK的结合,从而影响OPG/RANK/RANKL系统,进一步影响乳腺癌转移,有利于从分子机制上阐明紫甘薯多糖的抗肿瘤活性,为治疗癌转移和研发新药提供可能。
技术实现要素:
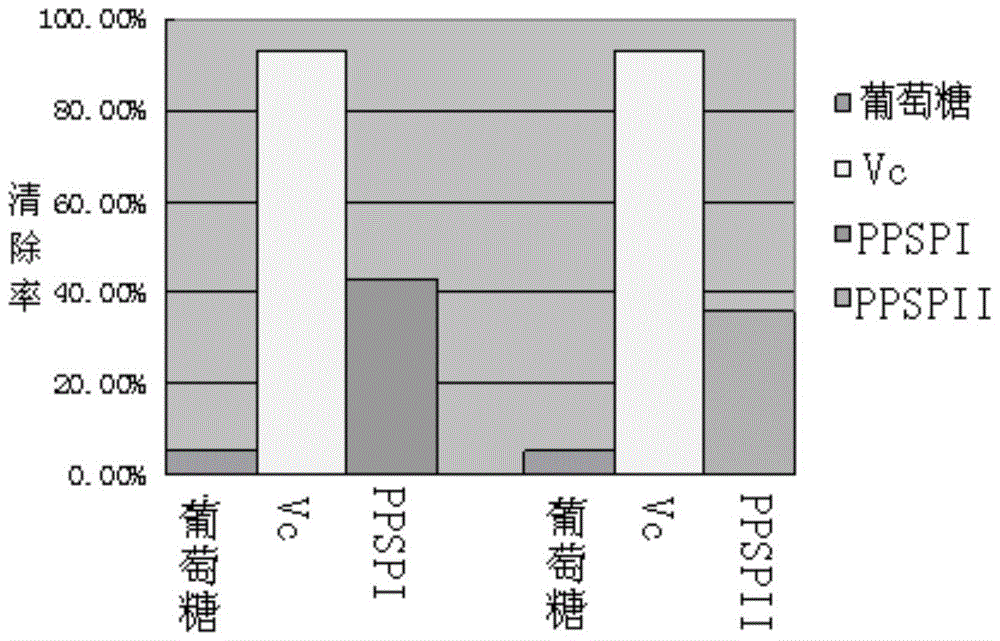
本发明的目的在于提供紫甘薯多糖及其提取方法。本发明的另一个目的是提供该紫甘薯多糖在制备抗氧化的功能性食品中的用途。为实现本发明所说的紫甘薯多糖的提取,采用的提取方法包括如下步骤:1).将紫甘薯烘干粉碎成粉末,颗粒直径为60~140目;2).将步骤1)得到的紫甘薯粉末,加入木聚糖酶,用8倍质量于紫甘薯粉末的水悬浮,在50℃~60℃下,160~180r/min震荡1小时,之后于75℃~80℃恒温水浴5~15min让酶失活,制成悬浮液Ⅰ;3).将步骤2)得到的悬浮液Ⅰ2000~4000r/min离心5~10min,取上清,向上清液中加入3倍体积的无水乙醇,0~4℃放置8小时以上,然后2000~4000r/min离心5~10min,取沉淀,将沉淀用蒸馏水制成悬浮液Ⅱ;4).向步骤3)得到的悬浮液Ⅱ中加入0.25倍体积的sevage液脱蛋白,反复洗脱3-5次,每次30min,充分振荡,静止30min或2000~4000r/min离心5~10min,取上层液体,即多糖水溶液Ⅰ;5).向步骤4)得到的多糖水溶液Ⅰ中加入活性炭脱色,脱色条件为振荡20~30min,4000~6500r/min离心8~10min,脱色完成后,以不低于12000r/min的转速离心8~10min取上清液,得到脱色后的多糖水溶液,即多糖水溶液Ⅱ;6).将步骤5)得到的多糖水溶液Ⅱ干燥,即得到紫甘薯多糖;其中,步骤4)所述的sevage液由4倍体积的氯仿和1倍体积的正丁醇混合而成。优选地,步骤2)所述加入的木聚糖酶质量是所述紫甘薯粉末质量的0.6%,活力单位为1~15万IU。优选地,步骤5)所述的活性炭的质量按照所述多糖水溶液Ⅰ质量的3%加入,用前要烘干数天。优选地,步骤5)所述的脱色为反复脱两次至溶液清亮透彻。优选地,在步骤6)中所述的将多糖水溶液Ⅱ干燥之前,将所述多糖水溶液Ⅱ用阴离子交换层析法纯化,用浓度为0.3~2M的NaCL溶液洗脱,流速控制在1.0ml/min,洗脱完成后去除NaCL,然后再进行所述的干燥。进一步优选地,所述洗脱用NaCL溶液的浓度为0.3~0.8M,所述去除NaCL的方法是透析法或色谱分离法。更进一步优选地,所述洗脱用NaCL溶液的浓度为0.5M。阴离子交换层析法优选地采用的是Cellulose阴离子交换层析柱,所述Cellulose阴离子交换层析柱用pH7.4、50mM磷酸缓冲溶液缓冲液平衡,平衡体积约为柱床体积的3~5倍,流速为1.5ml/min;所述Cellulose阴离子交换层析柱使用前还需要预处理,依次用含有0.2M氢氧化钠的8M尿素、含体积百分比为0.5%的tritonX-100的1M醋酸、体积百分比为70%乙醇、0.3M氢氧化钠淋洗。优选地,在步骤6)中所述的干燥为冷冻干燥或喷雾干燥。附图说明图1为PPSP的DEAECellulose阴离子交换层析图;图2为PPSPⅠ、PPSPⅡ、葡萄糖和Vc对羟自由基的清除率比较图;图3为RANKL蛋白经His-tagNi亲和层析纯化后的SDS-PAGE电泳图;图4为经DEAE柱纯化后PPSPⅡ、PPSPⅢ、PPSPⅣ与RANKL蛋白作用的内源荧光图;图5为经DEAE柱纯化后PPSPⅡ、PPSPⅢ、PPSPⅣ与RANKL蛋白作用的ANS荧光图;图6为PPSPⅡ与RANKL蛋白作用的内源荧光图谱;图7为PPSPⅡ与RANKL蛋白作用的ANS荧光图谱;图8为苯酚-硫酸法测糖含量图;图9为考马斯亮蓝法测蛋白含量图;图10为紫甘薯多糖PPSPⅡ的红外光谱图;图11为紫甘薯多糖PPSPⅡ分子排阻层析图谱。具体实施方式现结合实施例详细说明本发明的技术方案。应理解,以下实施例仅用于说明本发明而不用于限制本发明的范围。在不背离本发明精神和实质的情况下,对本发明步骤或条件所作的修改或替换,均属于本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。实施例1紫甘薯多糖的提取方法之一A).紫甘薯多糖的提取将紫甘薯去皮、切片、晾干、粉碎,得到紫薯粉,颗粒直径为80目;称量5g的紫甘薯粉末,按紫甘薯粉末质量的0.6%加入木聚糖酶,加40mLddH2O;55℃、160r/min于恒温摇床中培养1h,之后于75℃恒温水浴5min让酶失活,制成悬浮液Ⅰ;将悬浮液Ⅰ2500r/min离心6min取上清,向其中加入3倍体积的无水乙醇,4℃过夜,然后2500r/min离心6min,取沉淀,将沉淀用蒸馏水制成悬浮液Ⅱ;用氯仿和正丁醇按照体积比为4:1配制sevage溶液,加入0.25倍悬浮液Ⅱ体积的sevage液液脱蛋白,反复洗脱55次,每次30min,充分振荡,静置30min,取上层液体,即多糖水溶液Ⅰ;按照多糖水溶液Ⅰ质量的3%加活性炭到多糖水溶液Ⅰ中,振荡30min,6500r/min离心10min,反复脱两次至溶液透彻为止,12000r/min离心10min取上清液,得到脱色后的多糖水溶液,即多糖水溶液Ⅱ;将多糖水溶液Ⅱ用冷冻干燥法干燥,即得到紫甘薯多糖0.411克,经测定,纯度为90.7%,计算得率为8.22%。产物多糖鉴别:苯酚-硫酸试剂反应呈阳性。B).紫甘薯多糖抗氧化活性试验i).紫甘薯多糖清除羟自由基能力的测定用硫酸铁和双氧水产生羟自由基,以氧化水杨酸所得产物的吸光值表示羟自由基的多少。反应液中加入不同的测试样品200uL(质量百分比浓度0.21%),最后加入双氧水启动反应。测510nm处的吸光值,以(A0一A1)/A0×100%作为羟自由基的清除率,其中A0为对照,A1为含样品的吸光值。结果见表1。表1紫甘薯多糖清除羟自由基能力组分空白对照Vc紫甘薯多糖A5100.7560.6130.716清除率(%)018.925.29由表1可知,紫甘薯多糖对羟自由基的清除率为Vc的27.96%。ii).紫甘薯多糖对邻苯三酚自氧化的抑制作用的测定取试样100uL(质量百分比浓度为0.21%),0.1mol/L的Tris-HClbuffer(pH值8.2,含4mmol/L的EDTA)1.8ml和双蒸水1.0ml,放入同一试管中,25℃水浴保温10min,加入同样温浴的3mmol/L邻苯三酚(用10mmoL/L的HCl配制)100uL,迅速混合后每隔30s测325nm处的吸光值A325,得出A325随时间变化的曲线,以回归方程的斜率作为邻苯三酚的自氧化速率V(△A/min)。结果见表2。抑制率(%)=[(V对照-V样本)/V对照]×100%表2紫甘薯多糖对邻苯三酚自氧化的抑制作用组分对照紫甘薯多糖自氧化速率0.12030.1011抑制率(%)015.96由表2可知,实验提取的紫甘薯多糖对临苯三酚的抑制效果可达到至少15.96%。综合上述实验结果,本发明提供的紫甘薯多糖具有显著的抗氧化活性,可以用于制备保健药物和保健食品。实施例2紫甘薯多糖的提取方法之二将紫甘薯去皮、切片、晾干、粉碎,得到紫薯粉,颗粒直径为60目;称量5g的紫甘薯粉末,按紫甘薯粉末质量的0.6%加入木聚糖酶,加40mLddH2O;50℃、180r/min于恒温摇床中培养1h,之后于75℃恒温水浴8min让酶失活,制成悬浮液Ⅰ;将悬浮液Ⅰ2000r/min离心10min取上清,向其中加入3倍体积的无水乙醇,2℃过夜,然后2000r/min离心10min,取沉淀,将沉淀用蒸馏水制成悬浮液Ⅱ;用氯仿和正丁醇按照体积比为4:1配制sevage溶液,加入0.25倍悬浮液Ⅱ体积的sevage液液脱蛋白,反复洗脱55次,每次30min,充分振荡,2000r/min离心10min,取上层液体,即多糖水溶液Ⅰ;按照多糖水溶液Ⅰ质量的3%加活性炭到多糖水溶液Ⅰ中,振荡25min,5500r/min离心8min,反复脱两次至溶液透彻为止,13000r/min离心10min取上清液,得到脱色后的多糖水溶液,即多糖水溶液Ⅱ;处理阴离子交换柱,配8M尿素和氢氧化钠100ml打入柱内,接着将1M醋酸100ml打入柱内,再用70%乙醇洗,除掉色素,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜;用0.3MHCl40ml洗,蒸馏水洗过夜,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜,将适量处理好的阴离子交换Cellulose装柱,先用pH7.450mM磷酸缓冲溶液缓冲液平衡,平衡体积约为柱床体积的3~5倍,流速为1.5ml/min;将多糖水溶液Ⅱ上样,用0.3MNaCL溶液洗脱,流速控制在1.0ml/min;透析法去除NaCL,喷雾干燥法干燥得紫甘薯多糖0.112克,经测定,纯度为95.4%,计算得率为2.24%。多糖鉴别:苯酚-硫酸试剂反应呈阳性。实施例3紫甘薯多糖的提取方法之三将紫甘薯去皮、切片、晾干、粉碎,得到紫薯粉,颗粒直径为70目;称量5g的紫甘薯粉末,按紫甘薯粉末质量的0.6%加入木聚糖酶,加40mLddH2O;55℃、170r/min于恒温摇床中培养1h,之后于80℃恒温水浴5min让酶失活,制成悬浮液Ⅰ;将悬浮液Ⅰ3000r/min离心6min取上清,向其中加入3倍体积的无水乙醇,0℃过夜,然后3000r/min离心8min,取沉淀,将沉淀用蒸馏水制成悬浮液Ⅱ;用氯仿和正丁醇按照体积比为4:1配制sevage溶液,加入0.25倍悬浮液Ⅱ体积的sevage液液脱蛋白,反复洗脱55次,每次30min,充分振荡,4000r/min离心5min,取上层液体,即多糖水溶液Ⅰ;按照多糖水溶液Ⅰ质量的3%加活性炭到多糖水溶液Ⅰ中,振荡20min,6500r/min离心8min,反复脱两次至溶液透彻为止,12500r/min离心8min取上清液,得到脱色后的多糖水溶液,即多糖水溶液Ⅱ;处理阴离子交换柱,配8M尿素和氢氧化钠100ml打入柱内,接着将1M醋酸100ml打入柱内,再用70%乙醇洗,除掉色素,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜;用0.3MHCL40ml洗,蒸馏水洗过夜,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜,将适量处理好的阴离子交换Cellulose装柱,先用pH7.4、50mM磷酸缓冲溶液缓冲液平衡,平衡体积约为柱床体积的3~5倍,流速为1.5ml/min;将多糖水溶液Ⅱ上样,用0.5MNaCL溶液洗脱,流速控制在1.0ml/min;透析法去除NaCL,用喷雾干燥法干燥得到紫甘薯多糖0.208克,经测定,纯度为96.0%,计算得率为4.16%。多糖鉴别:苯酚-硫酸试剂反应呈阳性。实施例4紫甘薯多糖的提取方法之四将紫甘薯去皮、切片、晾干、粉碎,得到紫薯粉,颗粒直径为100目;称量5g的紫甘薯粉末,按紫甘薯粉末质量的0.6%加入木聚糖酶,加40mLddH2O;60℃、180r/min于恒温摇床中培养1h,之后于80℃恒温水浴8min让酶失活,制成悬浮液Ⅰ;将悬浮液Ⅰ2500r/min离心8min取上清,向其中加入3倍体积的无水乙醇,4℃过夜,然后4000r/min离心5min,取沉淀,将沉淀用蒸馏水制成悬浮液Ⅱ;用氯仿和正丁醇按照体积比为4:1配制sevage溶液,加入0.25倍悬浮液Ⅱ体积的sevage液液脱蛋白,反复洗脱55次,每次30min,充分振荡,3000r/min离心6min,取上层液体,即多糖水溶液Ⅰ;按照多糖水溶液Ⅰ质量的3%加活性炭到多糖水溶液Ⅰ中,振荡30min,5500r/min离心8min,反复脱两次至溶液透彻为止,12000r/min离心9min取上清液,得到脱色后的多糖水溶液,即多糖水溶液Ⅱ;处理阴离子交换柱,配8M尿素和氢氧化钠100ml打入柱内,接着将1M醋酸100ml打入柱内,再用70%乙醇洗,除掉色素,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜;用0.3MHCL40ml洗,蒸馏水洗过夜,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜,将适量处理好的阴离子交换Cellulose装柱,先用pH7.4、50mM磷酸缓冲溶液缓冲液平衡,平衡体积约为柱床体积的3~5倍,流速为1.5ml/min;将多糖水溶液Ⅱ上样,用0.8MNaCL溶液洗脱,流速控制在1.0ml/min;色谱分离法去除NaCL,用冷冻干燥法干燥得紫甘薯多糖0.295克,经测定,纯度为95.8%,计算得率为5.90%。多糖鉴别:苯酚-硫酸试剂反应呈阳性。实施例5紫甘薯多糖的提取方法之五将紫甘薯去皮、切片、晾干、粉碎,得到紫薯粉,颗粒直径为90目;称量5g的紫甘薯粉末,按紫甘薯粉末质量的0.6%加入木聚糖酶,加40mLddH2O;55℃、160r/min于恒温摇床中培养1h,之后于80℃恒温水浴5min让酶失活,制成悬浮液Ⅰ;将悬浮液Ⅰ4000r/min离心5min取上清,向其中加入3倍体积的无水乙醇,2℃过夜,然后2500r/min离心10min,取沉淀,将沉淀用蒸馏水制成悬浮液Ⅱ;用氯仿和正丁醇按照体积比为4:1配制sevage溶液,加入0.25倍悬浮液Ⅱ体积的sevage液液脱蛋白,反复洗脱55次,每次30min,充分振荡,静止30min,取上层液体,即多糖水溶液Ⅰ;按照多糖水溶液Ⅰ质量的3%加活性炭到多糖水溶液Ⅰ中,振荡20min,6500r/min离心8min,反复脱两次至溶液透彻为止,13000r/min离心10min取上清液,得到脱色后的多糖水溶液,即多糖水溶液Ⅱ;处理阴离子交换柱,配8M尿素和氢氧化钠100ml打入柱内,接着将1M醋酸100ml打入柱内,再用70%乙醇洗,除掉色素,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜;用0.3MHCl40ml洗,蒸馏水洗过夜,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜,将适量处理好的阴离子交换Cellulose装柱,先用pH7.4、50mM磷酸缓冲溶液缓冲液平衡,平衡体积约为柱床体积的3~5倍,流速为1.5ml/min;将多糖水溶液Ⅱ上样,用2.0MNaCL溶液洗脱,流速控制在1.0ml/min;透析法去除NaCL,用喷雾干燥法干燥得紫甘薯多糖0.397克,经测定,纯度为96.3%,计算得率为7.94%。多糖鉴别:苯酚-硫酸试剂反应呈阳性。实施例6紫甘薯多糖抗氧化活性试验A).紫甘薯多糖的提取和检测1).紫甘薯多糖的提取将紫甘薯去皮、切片、晾干、粉碎,得到紫薯粉,颗粒直径为60目;称量5g的紫甘薯粉末,按紫甘薯粉末质量的0.6%加入木聚糖酶,加40mLddH2O;60℃、160r/min于恒温摇床中培养1h,之后于75℃恒温水浴10min让酶失活,制成悬浮液Ⅰ;将悬浮液Ⅰ3000r/min离心5min取上清,向其中加入3倍体积的无水乙醇,0℃过夜,然后4000r/min离心5min,取沉淀,将沉淀用蒸馏水制成悬浮液Ⅱ;用氯仿和正丁醇按照体积比为4:1配制sevage溶液,加入0.25倍悬浮液Ⅱ体积的sevage液液脱蛋白,反复洗脱55次,每次30min,充分振荡,2500r/min离心8min,取上层液体,即多糖水溶液Ⅰ;按照多糖水溶液Ⅰ质量的3%加活性炭到多糖水溶液Ⅰ中,振荡30min,4000r/min离心10min,反复脱两次至溶液透彻为止,13000r/min离心10min取上清液,得到脱色后的多糖水溶液,即多糖水溶液Ⅱ;处理阴离子交换柱,配8M尿素和氢氧化钠100ml打入柱内,接着将1M醋酸100ml打入柱内,再用70%乙醇洗,除掉色素,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜;用0.3MHCl40ml洗,蒸馏水洗过夜,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜,将适量处理好的阴离子交换Cellulose装柱,先用pH7.4、50mM磷酸缓冲溶液缓冲液平衡,平衡体积约为柱床体积的3~5倍,流速为1.5ml/min;将多糖水溶液Ⅱ上样,分别用0.3M、0.5M、0.8M、2MNaCL洗脱,流速控制在1.0ml/min,每管10ml,分部收集,分别得到四个组分即:紫甘薯多糖组分Ⅰ(PPSPⅠ)、PPSPⅡ、PPSPⅢ、PPSPⅣ。2).紫甘薯多糖的检测(见图1)图1为用苯酚-硫酸法检测,以490nm处糖的吸收值为纵坐标,洗脱管数为横坐标绘制的洗脱曲线,由图可以看出有四个峰,与紫甘薯多糖粗提液经DEAECellulose阴离子交换柱纯化所得层析图谱的出峰情况基本一致,主要得到四个组分,分别为PPSPⅠ、PPSPⅡ、PPSPⅢ、PPSPⅣ,依次为0.3M、0.5M、0.8M、2MNaCL洗脱。B).紫甘薯多糖抗氧化活性试验i).紫甘薯多糖清除羟自由基能力的测定用硫酸铁和双氧水产生羟自由基,以氧化水杨酸所得产物的吸光值表示羟自由基的多少。反应液中加入不同的测试样品200uL,最后加入双氧水启动反应。测510nm处的吸光值,以(A0一A1)/A0×100%作为羟自由基的清除率,其中A0为对照,A1为含样品的吸光值。结果见表3。表3紫甘薯多糖清除羟自由基能力组分空白对照葡萄糖VcPPSPⅠPPSPⅡA5100.0970.0920.0070.0550.062清除率(%)05.1592.7843.3036.08其中PPSPⅢ和PPSPⅣ清除率为负值,不纳入表格。PPSPⅠ、PPSPⅡ、葡萄糖和Vc对羟自由基的清除率比较见图2由图2可知,实验提取紫甘薯多糖对羟自由基的清除率远远大于葡萄糖,并几乎可达到清除率相当高的Vc的一半,紫甘薯多糖的清除率为Vc的40-50%。ii).多糖对邻苯三酚自氧化的抑制作用的测定取试样100uL,0.1mol/L的Tris-HClbuffer(pH值8.2,含4mmol/L的EDTA)1.8ml和双蒸水1.0ml,放人同一试管中,25℃水浴保温10min,加入同样温浴的3mmol/L邻苯三酚(用10mmoL/L的HCl配制)100uL,迅速混合后每隔30s测325nm处的吸光值A325,得出A325随时间变化的曲线,以回归方程的斜率作为邻苯三酚的自氧化速率V(△A/min)。结果见表4。抑制率(%)=[(V对照-V样本)/V对照]×100%表4紫甘薯多糖对邻苯三酚自氧化的抑制作用组分对照PPSPⅠPPSPⅡPPSPⅢPPSPⅣ自氧化速率0.00590.00350.00140.00220.0034抑制率(%)040.6876.2762.7142.37由表4可知,实验提取的紫甘薯多糖对临苯三酚的抑制效果可达到至少40%。实施例7紫甘薯多糖潜在的抗肿瘤效能试验A).紫甘薯多糖的提取将紫甘薯去皮、切片、晾干、粉碎,得到紫薯粉,颗粒直径为80目;称量5g的紫甘薯粉末,按紫甘薯粉末质量的0.6%加入木聚糖酶,加40mLddH2O;55℃、180r/min于恒温摇床中培养1h,之后于80℃恒温水浴5min让酶失活,制成悬浮液Ⅰ;将悬浮液Ⅰ4000r/min离心6min取上清,向其中加入3倍体积的无水乙醇,4℃过夜,然后3000r/min离心5min,取沉淀,将沉淀用蒸馏水制成悬浮液Ⅱ;用氯仿和正丁醇按照体积比为4:1配制sevage溶液,加入0.25倍悬浮液Ⅱ体积的sevage液液脱蛋白,反复洗脱55次,每次30min,充分振荡,4000r/min离心6min,取上层液体,即多糖水溶液Ⅰ;按照多糖水溶液Ⅰ质量的3%加活性炭到多糖水溶液Ⅰ中,振荡30min,4500r/min离心9min,反复脱两次至溶液透彻为止,12000r/min离心10min取上清液,得到脱色后的多糖水溶液,即多糖水溶液Ⅱ;处理阴离子交换柱,配8M尿素和氢氧化钠100ml打入柱内,接着将1M醋酸100ml打入柱内,再用70%乙醇洗,除掉色素,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜;用0.3MHCl40ml洗,蒸馏水洗过夜,用0.3M氢氧化钠40ml洗20min,蒸馏水过夜,将适量处理好的阴离子交换Cellulose装柱,先用pH7.450mM磷酸缓冲溶液缓冲液平衡,平衡体积约为柱床体积的3~5倍,流速为1.5ml/min;将多糖水溶液Ⅱ上样,分别用0.3M、0.5M、0.8M、2MNaCL洗脱,流速控制在1.0ml/min,每管10ml,分部收集,分别得到四个组分即:紫甘薯多糖组分Ⅰ(PPSPⅠ)、PPSPⅡ、PPSPⅢ、PPSPⅣ。B).细胞核因子κB受体活化因子配体(RANKL)蛋白的提取与分离纯化1).活化菌种向6mlLB液体培养基中加入6μl50mg/ml氨苄霉素(工作浓度为50μg/ml),再接入100μl表达菌种,于37℃摇床振荡培养8-12小时。2).扩大培养向300mlLB液体培养基中加入300μl50mg/ml氨苄霉素(工作浓度为50μg/ml),再加入6ml活化的菌液,于37℃恒温培养箱中培养3小时。3).IPTG诱导加入IPTG(终浓度20mM)16℃160r/min过夜诱导。4).蛋白质提取将菌液置于离心管4℃,5000r/m离心10min;弃上清液,向沉淀中加入蒸馏水,用漩涡混合器混匀;4℃,5000r/m离心10min;弃上清,向沉淀中加入裂解液,用漩涡混合器混匀;在冰上进行10min(工作时间为2s,间隔为3s)的超声波破壁,一共三次,每两次间间隔5min;4℃,12000rpm,离心10min,取上清,得到RANKL的粗提液。5).蛋白质纯化上样:插A280滤光片,调T值(100)、A值(0);打开采集器;打开电脑蛋白核酸检测系统,保存文档并开始采集。将粗提液45ml上柱,流速控制约为1ml/min。洗脱:上样完后,继续用PBS缓冲液(pH=7.4)淋洗。待杂蛋白峰回落走平之后开始用不同梯度的咪唑(40mM、80mM、150mM、200mM)洗脱。在280nm处进行连续紫外检测,根据微电脑记录仪显示曲线。分别收集峰值的洗脱液根据波峰对每管收集蛋白测活并留样、电泳检测纯化程度。6).SDS-PAGE电泳检测(见图3)由图3知,第1孔为实验组上清,可以明显看到有一强带,为目标蛋白;第2孔中的穿流液为大量除目标蛋白以外的杂蛋白,强带所在的条线上,穿流比粗提液要浅一些,其余条带颜色的深浅基本上一致;第3至5孔为洗脱液,洗脱之后杂带大部分消失,目标蛋白的强带颜色较杂带更深。相比之下,40mM咪唑只洗脱下少量的目标蛋白,80mM咪唑洗脱的蛋白较纯,虽有少量杂带,但还是目标蛋白为主带,可用,150mM咪唑虽然洗脱下大量目标蛋白,但是杂带也很强,不可用。C).紫甘薯多糖抗潜在的抗肿瘤能力试验i).三个组分与RANKL蛋白作用的内源和外源荧光比较(见图4、图5)由图4和图5可以看出,PPSPⅡ、PPSPⅢ、PPSPⅣ三个组分相比,PPSPⅡ在内源和外源荧光中都对RANKL蛋白作用更明显一些,在内源图中,PPSPⅡ作用于RANKL蛋白后递减的趋势最强,表明其对RANKL蛋白的结构影响最强烈。在外源图中,PPSPⅡ的递增趋势也是最明显的,说明其对RANKL蛋白疏水面的影响最大,因此,本实验采用PPSPⅡ来进一步研究其对RANKL蛋白的作用。ii).PPSPⅡ与RANKL蛋白作用的内源荧光测定结果(见图6)图6表明随着PPSPⅡ浓度的增加,反应体系的荧光强度呈递减趋势,表明多糖与RANKL作用后对RANKL的结构产生了一定的影响。iii).PPSPⅡ与RANKL蛋白作用的ANS荧光测定结果(见图7)图7显示,随着PPSPⅡ浓度的增加,反应体系的荧光强度呈递增的趋势,表明RANKL蛋白疏水面变大,推测可能是多糖与RANKL作用后改变了其单体的构象,使其疏水面变大,也有可能是多糖与RANKL作用后使其三聚体的状态发生了改变,解聚成二聚体或单体。D).紫甘薯多糖PPSPⅡ的结构分析i).苯酚-硫酸法测糖含量(见图8)ii).考马斯亮蓝法测蛋白含量(见图9)由图8和图9表明:①PPSPⅡ中糖的含量为:10*(0.11-0.0022)/0.5445=1.9798(mg/ml);②PPSPⅡ中蛋白含量:20*(0.023-0.0161)/0.0108=12.7778(μg/ml)=0.0127778(mg/ml);③PPSPⅡ中糖与蛋白的比例为:1.9798/0.0127778=155,说明此组分中主要含糖,蛋白的含量很少,可能不是一种糖蛋白。iii).紫甘薯多糖PPSPⅡ的红外光谱分析(见图10)由图10分析知:842.6cm-1处的峰表明PPSPⅡ为a-D-葡萄吡喃糖(855~833);951.9cm-1处的吸收峰表示PPSPⅡ为吡喃糖(930~960);在1108.6cm-1附近有环内醚C-O伸缩振动(1150~1080);在1400~1200cm-1所看到的吸收峰为C-H的变角振动;在1464.6cm-1处存在次甲基C-H变角振动(1465);1643.1cm-1附近有N-H变角振动(1650~1500),无C=O伸缩振动(1740~1680);在3434.7cm-1附近有O-H伸缩振动的强吸收峰(3700~3100)。iv).紫甘薯多糖PPSPⅡ分子排阻层析图谱分析(AKTApurifier)(见图11)图11结果表明,多糖没有特征吸收峰,根据purifier图谱基本判断PPSPⅡ为较均一组分,糖苷链中存在着氨基酸。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1