一种人源溶菌酶蛋白发酵方法与流程
本发明属于基因工程和发酵工程领域,具体而言,本发明涉及一种发酵人源溶菌酶蛋白的生产工艺方法的应用。
背景技术:
感染性疾病一直是人类健康和生命的主要威胁,目前对于细菌性感染,抗生素及人工合成抗菌药物仍是最主要的治疗手段。然而进入21世纪后,不断出现的多重耐药细菌使感染性疾病的治疗困难重重,如甲氧西林耐药金黄色葡萄球菌(MRSA)、万古霉素耐药肠球菌(VRE)、青霉素耐药肺炎链球菌(PRSP)、泛耐药(PDR)铜绿假单胞菌、鲍曼不动杆菌等各种超级耐药细菌的出现。因此寻找和开发与传统抗生素结构与作用机制以及作用靶位不同的抗菌药物已是当务之急。
溶菌酶(Lysozyme)是一种水解酶,它能够水解细菌细胞壁中肽聚糖的糖苷键,又称胞壁质酶。该酶由129个氨基酸残基组成的碱性球蛋白,作用机理是切断肽聚糖中N-乙酰胞壁酸(NAM)与N-乙酰葡萄糖胺(NAG)之间的β-1,4-糖苷键之间的联结,破坏肽聚糖支架,在内部渗透压的作用下细胞胀裂开,引起细菌裂解。溶菌酶在自然界中分布广泛,按照溶菌酶的不同物种来源,可分为:鸡型(c-型,chicken type)、鹅型(g-型,goose type)、无脊椎动物型(i-型,invertebrate type)、噬菌体溶菌酶、植物溶菌酶和细菌溶菌酶。在人体中同时存在鸡型和鹅型,也就是常说的c-型和g-型两种溶菌酶,其中c-型溶菌酶的研究最为广泛,因为它是唯一同时在脊椎动物、无脊椎动物和原索动物中存在的。c-型溶菌酶是大部分脊柱动物(包括哺乳动物)产生的主要溶菌酶,是作为酶结构和功能研究模型的原型溶菌酶。
溶菌酶是一种具有杀菌作用的天然物质,它具有耐热、耐酸的理化特性,可溶于水、乙醇、油脂类溶剂,通过裂解细胞壁而达到裂解革兰氏阳性细菌的效果。对溶菌酶杀菌能力研究表明:极微量的溶菌酶即可杀灭溶壁微球菌、藤黄球菌、巨大芽孢杆菌、棒状杆菌、乳酸杆菌、细球菌、八迭球菌、葡萄球菌等自然界常见细菌。
目前市场上的溶菌酶产品大多以蛋清溶菌酶为主要原料,因为鸡蛋清溶菌酶的提取工艺相对成熟,价格便宜,如上世纪80年代初上市的溶菌酶肠溶片、含片,我国SFDA已下达13个鸡溶菌酶肠溶片生产文号、16个含片生产文号。鸡溶菌酶产品在广州、上海、杭州的用量较大,分别占中国市场本品的56.55%、14.93%和9.25%。除了应用在医药行业,鸡溶菌酶在日化行业也得到了应用,如口腔护理产品牙膏、漱口液、喷剂,化妆品等。
但是,鸡蛋清溶菌酶对于人体而言是一种异源蛋白,在蛋白质一级结构上与人溶菌酶有较大差异,因此,它具有明显的局限性。鸡溶菌酶用于过敏体质人群时,可引发过敏反应,严重者可导致死亡。另外,鸡溶菌酶在人体内会产生免疫原性与毒副作用,无法用于静脉用药,对于侵入机体内的细菌、病毒无法产生抑制和杀灭作用。由于鸡溶菌酶的局限性,人们开始更加关注人溶菌酶(human lysozyme,h-LYZ)的研究和开发。人溶菌酶是在人奶汁中发 现的抗菌剂之一,且广泛存在于人的体液、细胞和组织器官中,如脾脏,肺脏,肾脏,白细胞,血浆,唾液和眼泪等。我们可以从人奶、胎盘或尿液中少量提取人溶菌酶,但制备材料相当有限,制备成本非常高,因此在医药、日化、食品、饲料添加剂等行业都没得到广泛应用。人溶菌酶与其他来源的溶菌酶相比,有许多无法逾越的优势,如酶活性是蛋清溶菌酶的3倍,并且人溶菌酶来源于人体,和人体具有天然相容性,不会产生任何副作用和免疫原性。目前全球最大最著名的sigma-aldrich生物/化学试剂公司(http://www.sigma-aldrich.com)出售的基因工程人溶菌酶(活性≥100ku/mg),售价478.53元/g,价格昂贵,只供各类研究机构使用。此外,从生物材料提取溶菌酶有其固有缺陷,一是受生物材料来源限制,二是生物材料由于取自不同生物体,保鲜程度不一,难以保证不受病毒和其他有害物质污染,这会造成产品质量不稳定。溶菌酶巨大的市场前景(主要有饲料市场、食品添加剂市场、化妆品市场和人用医药市场)也预示着,研究与开发人源溶菌酶(h-LYZ)势在必行。
技术实现要素:
本发明要解决的技术问题是提供一种操作简便、成本低廉、产品纯度高的人溶菌酶发酵方法,以达到工艺周期短、收率高,易于线性放大至生产规模的目的。
为了解决上述技术问题,本发明构建毕赤酵母(Pichia pastoris)表达系统及由酿酒酵母α-因子信号肽引导表达具有天然N端h-LYZ的重组pPIC9k-LYC6分泌型毕赤酵母表达载体,获得具有稳定整合表达载体的多拷贝插入菌株。通过摇瓶培养和诱导表达,初步筛选出蛋白表达量和活性较高的菌株,然后,利用Design-expert软件,依据响应面分析法对发酵条件进行优化,最后,在15L发酵罐中进行初步发酵,同时确定了最佳诱导pH值。
本发明反复测试并优化了高细胞密度发酵的工艺研究。在菌体生长阶段,采用不同的甘油补料方式(周期补料、恒速流加、指数流加及反馈补料-恒溶氧(DO-stat)流加),进行高密度发酵;在甲醇诱导阶段分别共混流加甘油、山梨醇、甘露醇和乳酸,研究了不同混合碳源对P.pastoris生长和h-LYZ蛋白表达的影响;防止外源蛋白降解策略研究,主要考察在诱导后期补加胰蛋白胨是否能有效防止蛋白降解;最后,在50L和500L发酵罐中进行放大,h-LYZ活性最高分别为151600U/mL和127300U/mL,蛋白表达量最高1.65g/L,且批次具有较好的稳定性。
本发明提供了一种人源溶菌酶蛋白的大规模工业化发酵方法,将表达人源溶菌酶的种子发酵液加入发酵罐进行高密度发酵。
其中,表达人源溶菌酶的种子发酵液是通过人源溶菌酶的酵母表达体系构建、定点整合至酵母基因组以及小规模发酵获得。
所述的人源溶菌酶的核酸序列源自NCBI网站数据库登录号为NM_000239.2。
所述的人源溶菌酶的酵母表达体系构建是以源自NCBI网站数据库登录号为NM_000239.2为基础,人工合成了去除自身信号肽的人源溶菌酶基因序列,并依据酵母偏爱 密码子构建由酿酒酵母α-因子信号肽引导表达具有天然N端人源溶菌酶的重组分泌型毕赤酵母载体。
构建人源溶菌酶的分泌型毕赤酵母的载体为pPIC9k。
所述的定点整合是指将线性化重组质粒分别电转入GS115和/或SMD1168中。
毕赤酵母感受态细胞的电转化具体步骤为:取重悬在ice-cold 1M sorbitol中的酵母感受态细胞80μL(约1010cells/mL)与3μg的线性化DNA混合,冰上放置5min(注意:质粒的体积应控制在10μL以内,体积太大会影响转化效率)。将混合物加入ice-cold的0.2cm电转杯中。电转参数为:电压2000V,电容25uF,电阻200Ω,电转时间5ms。转化结束后,立即加入0.5mL的ice-cold 1M sorbitol混匀。从电转杯中将液体吸出,转移到Eppendorf管中,30℃静置1小时。然后加入1mL YDP培养基,30℃,振荡培养4小时,取适量菌液涂布于MD平板。同时转化空质粒pPIC9k作为表达的阴性对照。
Mut+和Muts转化子筛选的具体步骤为:挑取MD平板上的His+转化子200个,接种至含有200μL YPD的96孔板中,30℃,培养24小时。每孔取1μL菌液点样于MM和MD平板上。30℃,培养至白色菌落出现。对比观察,若在MD和MM平板上生长均良好的克隆即Mut+表型;若MD平板上生长良好而在MM平板上生长不良或不长的克隆即为Muts表型。
多拷贝转化子筛选的具体步骤为:重组质粒插入酵母基因组会发生多拷贝插入,一般概率为1~10%,高拷贝插入可能会伴随着高蛋白表达。利用G418不同浓度的抗性可筛选出相对应的拷贝数。取培养在96孔板中的不同克隆,每孔取1μL点在含G418浓度为0.5,1,2,3,4,5mg/mL的YPD平板上。30℃,培养72小时。
所述的人源溶菌酶的核酸编码序列可以如SEQ ID NO 5所示。
重组毕赤酵母的摇瓶表达具体步骤为:经PCR阳性验证的G418高抗性菌株,进行摇瓶诱导表达实验。挑取单菌落接种于装有25mL BMGY培养基的250mL摇瓶中,28~30℃/250~300rpm培养至OD600=2~6;室温下5000rpm离心5min,收集菌体,用等体积BMMY培养基重悬菌体,继续培养;每24h向培养基中添加100%甲醇至终浓度为0.5~1.0%;按不同时间点取样品1mL,12000rpm,离心2min,收集上清。待检测样品置于-20℃保存,分析目的蛋白的表达量和酶活性。
所述的高细胞密度发酵包括甲醇诱导、添加胰蛋白胨和甘油补料。
发酵罐高密度发酵采用BSM发酵培养基,接种5%的种子培养液。
所述的BSM发酵培养基的配方为:磷酸26.7mL,CaSO40.93g,K2SO418.2g,MgSO4.7H2O 14.9g,KOH 4.13g,甘油40g,PTM14mL,泡敌1mL;加水至1L定容;
其中,PTM1溶液的配方为:CuSO4.5H2O 6g,KI 0.08g,MnSO4.H2O 3g,Na2MoO4.2H2O0.2g,H3BO30.02g,ZnSO4.7H2O 20g,FeSO4.7H2O 65g,CoCl2.6H2O 0.916g,H2SO45mL,Biotin 0.2g,加水至1L定容。
高密度发酵时,将含重组人源溶菌酶的发酵液调pH为4.5-6.5。
接种摇瓶的具体步骤为:挑取平板单菌落接种到1L YPD培养基中,30℃,220r/min培养20~24小时,当OD600=2~6时,可准备接种于发酵罐。
高密度发酵培养的步骤为:发酵罐里BSM发酵培养基灭菌,冷却后用25%氨水调节pH5.5,将培养好的YPD种子培养液通过火圈法接入15L/30L全自动发酵罐,接种量5%,初始搅拌转速为250r/min,通气量为2vvm,生长阶段温度为30℃,设置溶氧(DO)-转速联动,转速可根据DO的变化而变化。通过调节搅拌转速和通气量以维持DO在20%左右。当BSM分批发酵培养基中的甘油耗尽(DO迅速上升)时,采用不同的补料方式:周期补料、恒速流加、指数流加及恒溶氧值(DO-stat)补加同等量的50%甘油。当菌体达到一定浓度后,停止补料。待甘油再次耗尽(DO反弹),饥饿半小时后,开始以变速流加的方式限制性的流加补料诱导培养基,诱导h-LYZ表达。诱导阶段温度为24℃,pH4.5。诱导培养基的补料速率根据DO和OUR来控制。
甲醇诱导阶段分别共混流加甘油、山梨醇、甘露醇和乳酸。
甘油补料方式包括周期补料、恒速流加、指数流加或者反馈补料-恒溶氧流加。
人源溶菌酶蛋白的高密度发酵阶段,诱导温度在22~24℃。
人源溶菌酶蛋白的高密度发酵阶段,诱导时间在84~90小时。
发酵罐高密度发酵采用BSM发酵培养基,接种5%的YPD种子培养液。
YPD培养基:酵母提取物10g/L,蛋白胨20g/L,121℃高压灭菌20min,冷却至60℃后加入20%葡萄糖至终浓度为2%(20%葡萄糖过滤除菌)。配制固体培养基时含2%琼脂。
BSM分批发酵培养基(1L):H3PO4(85%)26.7mL,CaSO40.93g,K2SO418.2g,MgSO4.7H2O 14.9g,KOH 4.13g,甘油40g,PTM14mL,泡敌1mL。
PTM1溶液(1L):CuSO4.5H2O 6g,KI 0.08g,MnSO4.H2O 3g,Na2MoO4.2H2O 0.2g,H3BO30.02g,ZnSO4.7H2O 20g,FeSO4.7H2O 65g,CoCl2.6H2O 0.916g,H2SO45mL,Biotin0.2g。
高密度发酵阶段,补加甘油的量为50%。每隔12小时补加0.5%的胰蛋白胨。
本发明的创新如下:
1.在15L发酵罐中研究四种不同的甘油补料方式(周期补料、恒速流加、指数流加和反馈补料-恒溶氧(DO-stat)流加)对P.pastoris生长阶段的影响,通过比较菌体生长时间、细胞干重、菌体生长速率以及对甘油得率,得到最优的甘油补料方式。结果表明,指数流加在最短时间内出现DO反弹,细胞干重为142g/L,最大生长速率为0.35h-1。
2在30L和5L发酵罐中研究了混合碳源流加共诱导表达h-LYZ,在甲醇诱导阶段分别以0.5g/(L·h)的速率缓慢共混流加甘油、山梨醇、甘露醇和乳酸,结果发现流加山梨醇细胞死亡率降低了2.5%,同时胞内AOX酶活性提高了32%,比仅以甲醇为碳源诱导下,酶活提高了18.9%。
3.50L和500L放大结果,h-LYZ活性最高分别为151600U/mL和127300U/mL,蛋白表达量最高达1.65g/L,且批次具有较好的稳定性。放大结果理想,为工业化生产提供了实际参考依据。
附图说明
图1是pPIC9k和pPIC9质粒图谱。pPIC9与pPIC9k的区别在于缺少Kanamycin抗性基因。
图2是H-LYZ与动物LYZ/α-乳清蛋白序列进行多重比对结果。星号显示的是序列上100%的保守区域,冒号显示保守替代,点显示非保守替代。除了α-乳清蛋白外,其它来源的溶菌酶序列均具有c-型溶菌酶典型的8个保守结构氨基酸残基Cys(C),同时,都存在2个酶催化位点Glu35和Asp52。
图3是阳性克隆摇瓶诱导72h后蛋白表达结果。M:蛋白Marker;1:抗1mg/mL G418LYC6-GS115重组菌株;2:抗2mg/mL G418LYC6-SMD1168重组菌株;3:抗2mg/mL G418LYC6-GS115重组菌株;4:抗3mg/mL G418LYC6-SMD1168重组菌株;5:抗3mg/mL G418LYC6-GS115重组菌株;6:抗4mg/mL G418LYC6-SMD1168重组菌株;7:抗4mg/mL G418。
图4是诱导温度和诱导时间交互作用对酶活的影响。
图5是诱导温度和装液量交互作用对酶活的影响。
图6是诱导时间和装液量交互作用对酶活的影响。
图7是15L发酵不同取样时间点(0、12、24、36、48、60、72、84h)的酶活性曲线。
图8是15L发酵罐中,不同诱导pH的酶活性。
图9是SDS-PAGE检测发酵上清液蛋白表达量。1:蛋白Marker,2:标准品(Sigma人溶菌酶),3:摇瓶的蛋白表达量,4-6:15L发酵罐的蛋白表达量(诱导pH为5.5,5.0和4.5)。
图10是不同甘油补加方式下细胞干重变化曲线。
图11是不同甘油补加方式下菌体比生长速率曲线。
图12是诱导阶段细胞干重变化曲线。
图13是诱导阶段菌体比生长速率曲线。
图14是混合碳源的添加对h-LYZ酶活的影响。
图15是混合碳源的添加对蛋白表达量的影响。M:蛋白分子量标准;Lane1:Sigma LYZ;2:诱导0h;Line3-8:诱导90h发酵上清液,其中3为摇瓶发酵,4-8分别为甲醇、甘油/甲醇、乳酸/甲醇、甘露醇/甲醇和山梨醇/甲醇诱导下的发酵液,上样量均为5μL。
图16是不用诱导时间下的酵母细胞活力曲线。
图17是不同诱导时间下的酵母胞内AOX酶活曲线。
图18是补加胰蛋白胨对酶活的影响。
图19是50L发酵罐中蛋白表达量。M:蛋白Marker,1:标准品:上样量1μg(Sigma人溶菌酶),2~9:诱导12、24、36、48、60、72、84、90h发酵上清液:上样量4μL。目标条带为14.4kDa。
图20是500L发酵在线检测图。
具体实施方式
本发明使用的材料和方法如下。
一、材料
1.1菌株
E.coli DH5α:本实验室保存。
Pichia pastoris:GS115,SMD1168,Invitrogen公司。
溶壁微球菌:本实验室保存。
1.2载体(参见图1)
pPIC9,毕赤酵母分泌表达载体,Invitrogen公司。
pPIC9k,毕赤酵母分泌表达载体,Invitrogen公司。
1.3引物
LYC6-F:5'-GTGACTCGAGAAAAGAGAGGCTGAAGC-3'Xho I(SEQ ID NO 1)
LYC6-R:5'-TACCGAATTCCTAGACACCGCAACCTTGGACG-3'EcoR I(SEQ ID NO 2)
表达载体通用引物:
5'-AOX:5'-GACTGGTTCCAATTGACAAGC-3'(SEQ ID NO 3)
3'-AOX:5'-GCAAATGGCATTCTGACATCC-3'(SEQ ID NO 4)
1.4主要试剂
1.5培养基及试剂的配制
1.5.1培养基配方
LB培养基:酵母提取物5g/L,蛋白胨10g/L,NaCI 5g/L,121℃高压灭菌20min(用于培养pPIC9/pPIC9k原核宿主菌DH5α时,加入Amp100μg/mL),4℃保存。配制固体培养基时含2%琼脂。
YPD培养基:酵母提取物10g/L,蛋白胨20g/L,121℃高压灭菌20min,冷却至60℃后加入20%葡萄糖至终浓度为2%(20%葡萄糖过滤除菌)。配制固体培养基时含2%琼脂。
BMGY培养基:酵母提取物10g/L,蛋白胨20g/L,3g/L K2HPO4,11.8g/L KH2PO4,加水至890mL,121℃灭菌20min,冷却至60℃后加入10×YNB 100mL(13.4g/L),500×生物素1mL(4×10-4g/L),甘油10mL。
BMMY培养基:酵母提取物10g/L,蛋白胨20g/L,3g/L K2HPO4,11.8g/L KH2PO4,加水至895mL,121℃灭菌20min,冷却至60℃后加入10×YNB 100mL(13.4g/L),500×生物素1mL(4×10-4g/L),甲醇5mL。
MD培养基:80mL水中加入琼脂糖2g,121℃高压灭菌20min,冷却至60℃后加入10×YNB 10mL(13.4g/L),10×葡萄糖10mL(20g/L),500×生物素0.2m(4×10-4g/L)。
MM培养基:90mL水中加入琼脂糖2g,121℃高压灭菌20min,冷却至60℃后加入10×YNB 10mL(13.4g/L),500×生物素0.2mL(4×10-4g/L),100%甲醇0.5mL。
10×YNB(13.4%的无氨基酸酵母氮源):134gYNB溶于1L蒸馏水,过滤灭菌,4℃保存。
500×B(0.02%生物素):20mg生物素溶于100mL蒸馏水,过滤灭菌,4℃保存。
1.5.2试剂的配置
50×TAE Buffer(pH8.5):Tris 242g,Na2EDTA·2H2O 37.2g,800mL去离子水,57.1mL的醋酸,充分搅拌,加去离子水定容至1L后,室温保存。
5×Tris-Glycine Buffer(SDS-PAGE电泳缓冲液):0.125M Tris,1.25M Glycine,0.5%(w/v)SDS。
5×SDS-PAGE Buffer:1M Tris(pH6.8)0.25mL,0.5M DTT 0.077g,SDS 0.1g,溴酚蓝0.005g,甘油0.5mL,加去离子水补足至1mL,室温保存。
考马斯亮兰染色液:G-2501g,乙醇400mL,冰醋酸100mL,加去离子水定容至l L。
脱色液:乙醇200mL,乙酸100mL,加去离子水定容至I L。
1M山梨醇:182mg山梨醇溶解于100mL双蒸水中,过滤除菌。
BSM分批发酵培养基(1L):H3PO4(85%)26.7mL,CaSO40.93g,K2SO418.2g,MgSO4.7H2O 14.9g,KOH 4.13g,甘油40g,PTM14mL,泡敌1mL。
PTM1溶液(1L):CuSO4.5H2O 6g,KI 0.08g,MnSO4.H2O 3g,Na2MoO4.2H2O 0.2g,H3BO30.02g,ZnSO4.7H2O 20g,FeSO4.7H2O 65g,CoCl2.6H2O 0.916g,H2SO45mL,Biotin0.2g。
补料生长培养基:50%(w/v)甘油(含12mL/L PTM1)。
补料诱导培养基:100%甲醇(含12mL/L PTM1),20%(w/v)的甘油溶液(含12ml/LPTM1),20%(w/v)的山梨醇溶液(含12ml/L PTM1),20%(w/v)的甘露醇溶液(含12ml/LPTM1),20%(w/v)的乳酸溶液(含12ml/L PTM1)。
二、主要仪器设备
三、生物信息途径与分析软件
National Center for Biotechnology Information(NCBI):
http://www.ncbi.nlm.nih.gov/
Expert Protein Analysis System(EXPAST):http://web.expasy.org/protparam/
ORF Finder(Open Reading Frame Finder):
http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi
DNAWorks(v3.2.2):http://helixweb.nih.gov/dnaworks/
Signal 4.1server:http://www.cbs.dtu.dk/services/SignalP/
Primer 5.0软件
ClustalW2软件
Design Expert 8.0.5软件
四、实验方法
4.1具有天然N端的h-LYZ的酵母表达体系构建
4.1.1目的基因的扩增
Homo sapiens lysozyme(LYZ)编码的mRNA序列(NCBI登录号:NM_000239.2),并且根据毕赤酵母密码子的偏好性,设计了成熟蛋白基因编码序列,该序列命名为LYC6,该基因序列由上海生工生物工程有限公司合成。以LYC6F、LYC6R引物,PCR扩增目的基因片段,得到在5'端加入Xho I识别位点及Kex2酶切信号,3'端加入EcoR I识别位点的LYC6基因片段。
PCR反应体系:
PCR扩增条件:
95℃,5min;95℃,30s,60℃,30s,72℃,1min(34cycles);72℃,10min;4℃,5min。
PCR产物经1%琼脂糖凝胶电泳检测后用GenClean柱式琼脂糖凝胶DNA回收试剂盒回收。
4.1.2重组表达质粒pPIC9-LYC6的构建
上一步回收产物和表达载体pPIC9分别用限制性内切酶Xho I和EcoR I进行双酶切,酶切反应体系如下:
37℃酶切4小时后,然后将酶切产物经1%琼脂糖凝胶电泳检测后用GenClean柱式琼脂糖凝胶DNA回收试剂盒回收。
回收的目的片段和pPIC9载体进行连接,连接反应体系如下:
16℃连接过夜。
取上面的连接产物10μL加至100μL大肠杆菌DH5α感受态细胞中,冰浴30min,然后42℃水浴热激90s,立即置于冰浴3min。向每管混合物中加入0.5mL LB液体培养基,混匀后,37℃轻摇1h。5000rpm离心1min,弃上清,剩约100μL,将菌体重悬后涂布于含有100μg/mLAmpicillin的LB平板上,置于37℃培养箱中培养至长出白色透明单菌落。挑选单克隆,于1mL含有100μg/mL Ampicillin的LB液体培养基中,37℃震荡培养12h。然后菌液进行PCR验证,挑选出阳性克隆。
PCR反应体系:
PCR扩增条件:
95℃,5min;95℃,30s,56℃,30s,72℃,1min(30cycles);72℃,10min;4℃,5min。
PCR产物经1%琼脂糖凝胶电泳检测,阳性克隆鉴定后用GenClean柱式琼脂糖凝胶DNA回收试剂盒回收,同时菌液用终浓度15%无菌甘油保存于-20℃。
4.1.3重组表达质粒pPIC9K-LYC6的构建
上一步回收产物和表达载体pPIC9K分别用限制性内切酶BamH I和EcoR I进行双酶切,酶切反应体系如下:
37℃酶切4小时后,将酶切产物经1%琼脂糖凝胶电泳检测,然后用GenClean柱式琼脂糖凝胶DNA回收试剂盒回收。
回收的目的片段和pPIC9K载体进行连接,连接反应体系如下:
16℃连接过夜。
取上面的连接产物转化至大肠杆菌DH5α感受态细胞中(方法同上)。
用pPIC9K载体的通用引物5’AOX1和3’AOX1进行PCR鉴定(方法同上),鉴定出的阳性克隆菌液进行测序,测序结果利用Clustal W软件进行比对。比对结果正确的阳性克隆,甘油进行保菌,用于后续实验。
4.1.4重组质粒电转化毕赤酵母
将测序正确的克隆,扩大培养后提取质粒,方法参照《高纯度质粒小提中量试剂盒》说明。
首先将重组表达质粒pPIC9K-LYC6用Sal I单酶切,在His4位点进行线性化。酶切反应体系如下:
37℃酶切6小时后,取3μL产物用1%琼脂糖凝胶电泳检测是否切开。若完全切割后,进行下步操作。
采用苯酚、氯仿、异戊醇抽提法对线性化的质粒进行纯化浓缩,具体步骤为:取酶切后产物100μL,加入50μL苯酚和50μL氯仿/异戊醇(24:1),充分混匀,12000rpm离心5min。将上层无色液体转移到另一离心管中,加入1/10体积的醋酸钠缓冲溶液(pH=5.2),再加入2.5倍体积乙醇,-20℃放置30min,12000rpm离心10min。弃上清,用70%乙醇将沉淀混匀,再离心。弃上清,室温放置风干后用20μL无菌水溶解,-20℃保存备用。
毕赤酵母感受态细胞的制备:挑取酵母单菌落接种于50mLYPD培养液中,30℃振荡培养至OD600=1.0~2.0。室温,5000rpm离心5min,收集菌体。8×108的细胞重悬于10mL溶液A(100mM LiCl,10mM DTT,0.6M sorbitol和10mM Tris-HCl,pH 7.5)中,室温静置30min。4℃,5000rpm离心5min,收集沉淀,然后用1.5mL的ice-cold 1M sorbitol重悬,离心,收集沉淀。重复以上步骤三次。最后用500μL的ice-cold 1M sorbitol重悬,置冰上备 用。
毕赤酵母感受态细胞的电转化:取重悬在ice-cold 1M sorbitol中的酵母感受态细胞80μL(约1010cells/mL)与3μg的线性化DNA混合,冰上放置5min(注意:质粒的体积应控制在10μL以内,体积太大会影响转化效率)。将混合物加入ice-cold的0.2cm电转杯中。电转参数为:电压2000V,电容25uF,电阻200Ω,电转时间5ms。转化结束后,立即加入0.5mL的ice-cold 1M sorbitol混匀。从电转杯中将液体吸出,转移到Eppendorf管中,30℃静置1小时。然后加入1mL YDP培养基,30℃,振荡培养4小时,取适量菌液涂布于MD平板。同时转化空质粒pPIC9k作为表达的阴性对照。
4.1.5转化子的筛选
Mut+和Muts转化子的筛选:挑取MD平板上的His+转化子200个,接种至含有200μL YPD的96孔板中,30℃,培养24小时。每孔取1μL菌液点样于MM和MD平板上(注意:克隆编号一致)。30℃,培养至白色菌落出现。对比观察,若在MD和MM平板上生长均良好的克隆即Mut+表型;若MD平板上生长良好而在MM平板上生长不良或不长的克隆即为Muts表型。
多拷贝转化子的筛选:重组质粒插入酵母基因组会发生多拷贝插入,一般概率为1~10%,高拷贝插入可能会伴随着高蛋白表达。利用G418不同浓度的抗性可筛选出相对应的拷贝数。取培养在96孔板中的不同克隆,每孔取1μL点在含G418浓度为0.5,1,2,3,4,5mg/mL的YPD平板上。30℃,培养72小时。
4.2重组毕赤酵母的摇瓶表达
经PCR阳性验证的G418高抗性菌株,进行摇瓶诱导表达实验。挑取单菌落接种于装有25mL BMGY培养基的250mL摇瓶中,28~30℃/250~300rpm培养至OD600=2~6;室温下5000rpm离心5min,收集菌体,用等体积BMMY培养基重悬菌体,继续培养;每24h向培养基中添加100%甲醇至终浓度为0.5~1.0%;按不同时间点取样品1mL,12000rpm,离心2min,收集上清。待检测样品置于-20℃保存,分析目的蛋白的表达量和酶活性。
发酵上清液的蛋白检测:SDS-PAGE鉴定重组蛋白的表达。
发酵上清液的酶活分析:将紫外可见分光光度计调至450nm波长,底物悬浮液与酶液置于25℃下,吸取2.5mL底物悬浮液与0.1mL待测酶液加到1cm比色池中,迅速混匀计时,每隔5s测1次OD450;空白值用浓度为0.1mol/L磷酸盐缓冲液(pH 6.4)[11]。
酶活力单位的定义:室温25℃、pH 6.2时,波长450nm处,每分钟引起吸收度下降0.001为1个酶活力单位,那么每毫升发酵上清液中溶菌酶的酶活单位U/mL为:
式中:△OD450为75s与15s时的反应液吸光度值的差值。
4.3种子摇瓶培养
挑取平板单菌落接种到1L YPD培养基中,30℃,220r/min培养20~24小时,当OD600=2~6时,可准备接种于发酵罐。
4.3.1高密度发酵培养
发酵罐里BSM发酵培养基灭菌,冷却后用25%氨水调节pH5.5,将培养好的YPD种子培养液通过火圈法接入15L/30L全自动发酵罐,接种量5%,初始搅拌转速为250r/min,通气量为2vvm,生长阶段温度为30℃,设置溶氧(DO)-转速联动,转速可根据DO的变化而变化。通过调节搅拌转速和通气量以维持DO在20%左右。当BSM分批发酵培养基中的甘油耗尽(DO迅速上升)时,采用不同的补料方式:周期补料、恒速流加、指数流加及恒溶氧值(DO-stat)补加同等量的50%甘油。当菌体达到一定浓度后,停止补料。待甘油再次耗尽(DO反弹),饥饿半小时后,开始以变速流加的方式限制性的流加补料诱导培养基,诱导h-LYZ表达。诱导阶段温度为24℃,pH4.5。诱导培养基的补料速率根据DO和OUR来控制。发酵过程由发酵罐控制系统软件进行在线控制和数据采集。
4.3.2测定方法
4.3.2.1菌体干重的测定
菌液稀释后于波长600nm处以去离子水为对照进行比色测定,OD600=OD读数×稀释倍数。取10mL发酵液置于离心管中,10000r/min离心10min,弃上清,菌体用10mL无菌水重悬,再次离心。将离心后菌体置于80℃烘箱内,烘至恒重,称重并计算菌体干重(DCW,dry cell weight,单位g/L)。光密度值与细胞干重呈线性关系:Y=0.569X+0.492(X:OD600,Y:干重DCW)
4.3.2.2细胞活力的测定
检测原理:ABER活细胞传感仪的探头会发射电脉冲信号,在探头周围形成一个电磁场。活细胞有一个紧密凑合的细胞膜,在这个电磁场下可以被看作为一个独立的小电容器。由于电磁的影响,它会被极化,细胞膜周围的电荷会上升。而这个电容变化值会被ABER准确检测出来。而死细胞(膜破裂),起泡或是细胞碎片因不能形成一个密闭的电容,所以ABER将不会检测到。
取一系列同等体积处于对数生长期的菌液(细胞存活率100%),做OD-电容标准曲线。测得的电容值与光密度存在良好的线性关系:
Y=0.1948X+6.5009(X:OD600,Y:电容值)
4.3.2.3AOX醇氧化酶活力的测定
AOX催化生成的H2O2与4-氨基安替比邻(4-AAP)反应生成红色的醌,醌在490nm具有最大吸收光[1]。取50mL发酵液,5000r/min离心5min,弃掉上清液,沉淀用无菌水洗涤一次,再离心。将菌体重悬于0.1moL磷酸盐缓冲液(pH7.0)中,稀释至OD≈50,用高压细胞破碎仪在1000bar的压力下破碎酵母细胞,破碎时间5min。收集流出液,然后5000r/min离心5min。离心后的上清液中即含有胞内醇氧化酶AOX。
反应体系1mL:0.1M磷酸盐缓冲液(pH 7.0),0.4mM 4-AAP,25mM PSA,2U/mL HRP,25μL乙醇和25μLAOX酶液样品。在490nm的吸光值,25℃,反应1min,记录吸光度值的变化,进而计算出AOX的酶活(ε490nm=5.56mM-1cm-1)。
酶活定义:每分钟生成1μmoL的H202需要的醇氧化酶AOX的量。
3.2.5甲醇,甘油,山梨醇,甘露醇,乳酸浓度的测定
甲醇残留浓度测定:采用气相色谱仪测定与调控(GC-1120,East China University of Science and Technology)。色谱柱PEG20M,30m×0.32mm×0.50μm,毛细管柱。气化室:200℃,检测器(FID):220℃,柱箱:170℃,流速:N240mL/min,空气450mL/min,H240mL/min。
甘油,山梨醇,甘露醇,乳酸残留浓度测定:采用高效液相色谱仪测定与调控(LC1620,East China University of Science and Technology)。Cl8反相柱,5μm,4.6mm×250mm。流动相:70%乙腈,30%水。柱温:30℃,进样量:10μL,流速:0.6mL/min。检测器:示差折光检测器。
实施例1人源c-型溶菌酶的生物信息学分析 Homo sapiens lysozyme(LYZ)编码的mRNA序列源于NCBI登录号:NM_000239.2。优选的序列如SEQ ID NO 5所示。人源c-型溶菌酶(LYZ)与部分动物c-型溶菌酶/a-乳清蛋白的保守位点分析结果见图2。人源c-型溶菌酶与部分动物c-型溶菌酶/a-乳清蛋白的等电点分析见表1。
用ExPasy预测H-LYZ和动物LYZ/a-乳清蛋白的成熟蛋白等电点和分子量。预测结果,人源c-型溶菌酶蛋白的PI为9.28,a-乳清蛋白较其它蛋白的PI低,牛溶菌酶蛋白的PI偏中性,其余的溶菌酶均偏碱性,其中火鸡溶菌酶蛋白的PI最高。
表1 H-LYZ与动物LYZ/a-乳清蛋白的等电点和分子量预测结果
实施例2LYZ基因在毕赤酵母中的表达
2.1表达载体pPIC9K-LYC6的构建
用pPIC9K载体的通用引物5’AOX1和3’AOX1进行PCR鉴定阳性克隆,阳性克隆的大小约890bp,空载体约460bp,具体结果如图1所示。
pPIC9K-LYC6(LYC6即LYZ)重组质粒线性化后电转入毕赤酵母,能在不含组氨酸的MD板上长出白色的单菌落。挑取单菌落进行PCR验证,以LYC6-F和LYC6-R为引物,约在430bp出现特异性条带即为阳性克隆,说明目的基因已整合入毕赤酵母基因组中。
转化子的表型筛选结果,转化子几乎都是Mut+表型,与质粒整合方式预期结果一致。将96孔板中生长均一的克隆依次点在含G418浓度逐渐递增的YPD平板上,大多数通过转化重组Ppic9k而得到的His+重组子只含有单拷贝插入,但根据不同的抗性对应相应的拷贝数,可以筛选到高拷贝的转化子。
2.2摇瓶中蛋白表达和酶活检测结果
外源基因整合数量的不同可能会造成蛋白表达量的差异,尽管有文献报道,高拷贝和蛋白表达量成正比,但拷贝数并不是越高越好,可能原因是外源mRNA的翻译效率或者蛋白在内质网中不能正确折叠,所以合适的拷贝数是需要的。实验中,经PCR验证为阳性的克隆,进行摇瓶诱导表达,进一步选择高产量和高活性的菌株。实验结果发现,溶菌酶活性和蛋白表达量存在正相关性。拷贝数和蛋白表达量存在一定的正相关性,其中筛选的LYC6-SMD1168G418高抗性克隆(3mg/mL)、LYC6-GS115G418高抗性克隆(3mg/mL)和LYC6-GS115G418高抗性克隆(4mg/mL)在摇瓶中的表达量较高。其中LYC6-GS115G418高抗性克隆(5mg/mL)蛋白条带异常,可能发生了错误折叠。
图3是抗4mg/mL G418LYC6-GS115重组菌株进行摇瓶诱导表达结果图,每隔12h取样,直至发酵结束(84h)。SDS-PAGE和酶活性分析结果,蛋白表达量和酶活性随着时间逐渐增加,在72h达到最高峰1510U/mL,之后随着诱导时间延长,蛋白量和活性无明显增长。
实施例3响应面分析法优化发酵条件结果
3.1Plackett-Burman设计法筛选重要因素
表2 Plackett-Burman试验设计与结果
对上述结果进行t检验,分析各因素的主效应,结果见表3。P值(Prob>F)的大小表明模型及各个考察因素的显著水平,七个因素对溶菌酶产量影响的显著性排列为:X4>X5>X2>X7>X1>X3>X6(诱导温度>诱导时间>装液量>甲醇诱导量>接种量>菌体生长时间>初始pH值)。其中X2,X4和X5(装液量,诱导温度和诱导时间)有显著性影响(P<0.01),作为进一步优化的因素。
表3 各因素影响的主效应分析
3.2响应面分析法确定因素的最佳水平
由Plackett-Burman实验结果可知:装液量,诱导温度和诱导时间对响应值的作用突出,因此这三个因子被Box-Behnken响应面分析其相互作用和最佳水平。对该3个因子重新编码,A(诱导温度)、B(诱导时间)和C(装液量),具体实验设计和结果见下表4。
由Design-Expert8.0.5软件拟合得多项式回归模型为:
Y=3194+228.56A+158.56B+175.75C+368.13AB-309.25AC+420.25BC-677.19A2-555.19B2-216.06C2
其中,Y代表预测的溶菌酶活性,A,B和C分别代表诱导温度,诱导时间和装液量的值。
对上述回归模型进行方差分析(表5),结果表明,模型Prob>F值小于0.0001,表明该模型是高度显著的。A,B,C,AB,AC,BC,A2,B2,C2为模型的显著因素(Prob>F值小于0.05)。模拟失拟项(Lack of Fit)表明模型预测值与实际值不拟合的概率,表1.7中Lack of Fit值为0.51,模型失拟项的Prob>F值为0.6990即方程模型失拟不显著,模型选择合适。信噪比Adeq Precision的值很高(为23.471),即该模型可用于预测;且Pred R-Squared值与Adj R-Squared值相差很小,亦表明该响应面方程不需要做进一步的优化。同时,软件分析得到模型的相关系数R2为98.09%,大于90%,说明模型相关度很好。变异系数(CV)反映模型的置信度,CV值越低模型的置信度越高[14],本实验的CV值为3.69%,说明模型方程能够很好地反映真实的实验值。所以,我们可以使用该模型来分析响应值的变化。回归方程一次项的回归系数绝对值一次为X1、X3、X2,这三个因素对发酵产溶菌酶影响大小依次为:诱导温度>装液量>诱导时间。
表4 Box-Behnken试验设计与结果
表5 响应面方差分析(ANOVA)
由Design-Expert8.0.软件绘制响应面和等高线(图4-6),进一步研究相关变量之间的相互作用及确定最优点。图4-6分别显示了三组以溶菌酶活性为响应值的趋势图,从等高线图可以直观地反映出两变量交互作用的显著程度,圆形表示两因素交互作用不显著,而椭圆形与之相反。图4-6a中等高线均呈椭圆形,表明两因素交互作用显著。从图4a可以看出,当诱导时间不变,随着诱导温度的增加,酶活先增大,当诱导温度超过23℃后,酶活呈逐渐下降的趋势。诱导温度在22~24℃,诱导时间在84~90h范围内,酶活较高。从图5b可见,当诱导温度 不变,随着装液量的增加,酶活也逐渐增加。毕赤酵母发酵过程消耗氧气,所以溶氧是一个重要的因素。摇瓶发酵中,提高转速或者减少装液量,均可以提高溶氧。但是在本研究中发现,酶活不是随着装液量的减少而提高,反而降低,存在的原因可能是甲醇沸点低,易挥发的特性,溶氧太高导致甲醇部分挥发掉,酵母没有足够的甲醇作为碳源,蛋白不能诱导表达。从图6可见,当装液量不变,随着诱导时间的增加,酶活先增大然后减小,在84h左右活性最高。温度升高,有利于菌体生长代谢和目的蛋白表达,但温度过高反而会使菌体过早衰老,发酵周期缩短,降低蛋白表达量和活性,因此,必须选择合适的温度和诱导时间,在不同阶段,选择不同温度,采用变温发酵策略。毕赤酵母菌体生长最适温度为30℃,诱导表达的温度应降低,常为20~28℃,尤其是分泌蛋白,因为低温可以降低折叠压力,有利于新生肽链的正确折叠。从各图可以看出A,B和C存在极值点,利用Design-Expert软件得知,三个因素的最优试验点(A,B,C)即(23.36℃,89.76h,47.89mL),此时Y最大值3315U/mL。考虑到实际发酵条件,各因素修正为:诱导阶段温度24℃,诱导时间90h,装液量48mL。该条件下对实验结果进行验证,溶菌酶的酶活为3301U/mL,实验值与理论预测值相对误差为0.42%,证明该模型是合理有效的,较优化前的酶活产量提高了2.2倍。
实施例415L发酵罐发酵结果
为了验证响应面优化结果,在15L的发酵罐进行了初级放大,为中试发酵提供参考依据。由于在摇瓶发酵过程中诱导阶段的pH值无法准确控制,所以在15L发酵罐中,采用单因素水平实验,摸索最适pH值。菌体生长阶段的pH控制在5.5,温度30℃,诱导阶段pH设3个点,分别是4.5,5.0和5.5,温度24℃。溶氧OD通过转速和空气通气量控制在20%作用,每隔8h取样。
发酵结果表明,诱导pH在4.5时,无论是蛋白表达量还是酶活性均最高,在诱导88h后,活性达到47680U/mL。SDS-PAGE分析摇瓶和15L发酵罐里的上清液(图7),结果显示在14.7kDa有目的性条带,目的蛋白达到总蛋白量的80%以上,为后续纯化工作带来便利。根据标品蛋白上样量1μg,估算摇瓶中溶菌酶蛋白表达量为0.2g/L~0.25g/L,发酵罐中表达量为0.8g/L~1.0g/L。
实施例5重组毕赤酵母高密度培养甘油补加策略的研究
毕赤酵母在BSM分批发酵培养阶段甘油的添加量由40g/L变为10g/L,DO在10小时之后降至20%,当生长12小时后DO反弹,说明毕赤酵母利用完BSM中的碳源。之后的甘油分批补料阶段分别采用:周期补料、恒速流加、指数流加及反馈补料四种方式来补加同等量的50%甘油。具体的操作步骤如下:
1)周期补料:以DO的上升为补料时间点,将补料培养基分成4等份,出现DO反弹后补加一次甘油。
2)恒速流加补料:是指以一定的速度连续补料,按照经验值,设置补料速度在8mL/L/h。
3)指数流加补料:以指数方式流加甘油,使细胞以一定的比生长速率呈指数形式增加。它是根据指数方程,从而计算出流速来补加甘油,常设定μPTI为0.16~0.18h-1[2]。流加速率F的计算公式:
式中X和S分别为细胞和底物浓度(g/L),μ为比生长速率(h-1),V为发酵液体积(L),SF为补加底物的浓度(g/L),YX/S为细胞对底物的得率系数(g/g),(VX)0为培养体系的初始细胞量(g),t为流加时间(h)。
表6 指数流加模型中的参数设定值
F(t)是时间的指数函数,实验中每2h阶梯式改变流加速率。整个补加过程采用流速先递增再递减的流加方式。
4)反馈补料-恒DO(也称作DO-stat):通过调整搅拌器使溶氧浓度维持在30%左右,补加甘油,维持在此数值,如果DO值升高,提高甘油补料速度;如果DO值下降,降低甘油补料速度。
1.1不同甘油补料方式对菌体生长时间和细胞干重的影响
四种不同的甘油补料方式(周期补料、恒速流加、指数流加及DO-反馈补料),毕赤酵母生长时间和细胞干重各不相同,按照周期补料方式进行甘油的补加,菌体生长36.5h后出现DO反弹,细胞干重为112g/L;恒速流加方式下菌体生长时间最长,为39.5h,DO反弹时细胞干重为126g/L;按照指数流加方式补加,菌体生长时间最短,为32h,DO反弹时细胞干重为142g/L;DO-star流加方式补加甘油,毕赤酵母在生长36h后出现DO反弹,细胞干重最高144g/L。
1.2不同甘油补料方式对菌体生长速率的影响
按照周期补料方式补加甘油,毕赤酵母在12h和20h出现两个最高比生长速率点,分别是0.25h-1和0.26h-1;恒速流加方式下,菌体在16h时比生长速率最高,为0.27h-1;按照指数流加方式补加甘油在16h时比生长速率最高,为0.35h-1;按照DO-star反馈流加方式补加甘油,在16h时出现最高比生长速率,为0.32h-1。其中,恒速流加、指数流加和DO-star流加三种方式均在同一时间段出现最高比生长速率。
表7 不同补料方式对P.pastoris生长的影响
Table.2.2Comparison of the influence of different culture modes on cell growth
实施例6混合碳源流加共诱导表达h-LYZ的研究
为了进一步提高重组毕赤酵母生产h-LYZ的产量,本实验在甲醇诱导阶段,采用多种碳源与甲醇混合添加的模式,以0.5g/(L·h)的速率缓慢共混流加甘油、山梨醇、甘露醇和乳酸,在24℃低温诱导表达h-LYZ。甲醇的流加速率根据《Pichia Fermentation Process Guidelines》作为依据,先以3.6mL/(L·h)的速率诱导2~3h,使P.pastoris适应甲醇(碳源从甘油到甲醇的转换阶段),之后逐渐地增加甲醇流速到7.3mL/(L·h),在诱导表达后期(70~100h)甲醇的流速调至10.9mL/(L·h)直至发酵结束。在诱导初期,为了防止当存在除甲醇之外其它碳源的时候,P.pastoris不能充分利用甲醇的现象,实验中我们采用甲醇诱导3h之后,再混合流加其余的碳源。
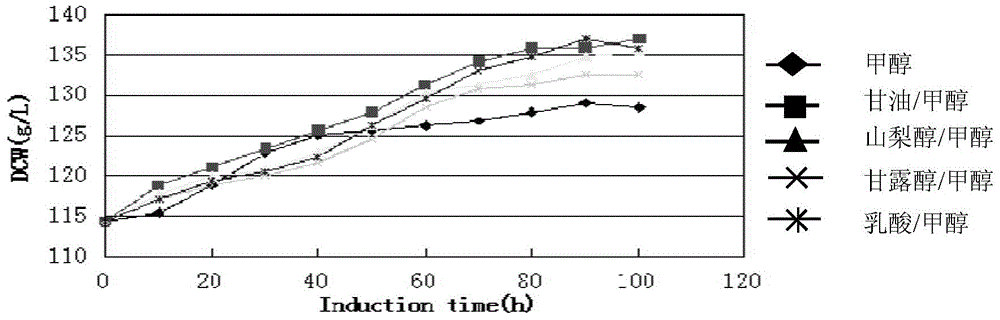
2.1混合碳源的添加对P.pastoris生长的影响
在甲醇诱导前细胞的干重均为114.3g/L,不同混合碳源在24℃下诱导100h后,细胞干重发生了明显的变化。只有甲醇存在的情况下,诱导结束后细胞干重为128g/L,平均比生长速率为0.0011h-1;甘油/甲醇共混诱导下,发酵结束时细胞干重为137.1g/L,平均比生长速率为0.0017h-1;山梨醇/甲醇共混诱导表达时,诱导结束后细胞干重为135.4g/L,平均比生长速率为0.0016h-1;甘露醇/甲醇共混诱导下,发酵结束时细胞干重为132.54g/L,平均比生长速率为0.0014h-1;乳酸/甲醇共混诱导表达时,诱导结束后细胞干重为135.9g/L,平均比 生长速率为0.0016h-1。
2.2混合碳源的添加对h-LYZ产量的影响
仅以甲醇为碳源,在诱导90h后酶活最高,为19380U/mL;甘油/甲醇共混诱导下,在诱导100h后酶活最高,为17800U/mL;山梨醇/甲醇共混诱导表达时,90h酶活最高值23900U/mL;甘露醇/甲醇共混诱导下,在90h酶活最高为23380U/mL;乳酸/甲醇共混诱导表达时,在90h酶活最高为23770U/mL。结果发现,山梨醇/甲醇共混合碳源诱导表达h-LYZ酶活最高。
2.3混合碳源的添加对细胞活力的影响
不同碳源混合诱导策略对细胞的活力影响不同。诱导50h时,细胞死亡率分别是:甲醇诱导条件下为16%;甘油/甲醇诱导条件下为14%;山梨醇/甲醇诱导条件下为13.5%;甘露醇/甲醇诱导条件下为13.5%;乳酸/甲醇诱导条件下为14%。发酵结束后,也就是发酵100h时,细胞死亡率分别是:甲醇诱导条件下为51%;甘油/甲醇诱导条件下为42%;山梨醇/甲醇诱导条件下为33%;甘露醇/甲醇诱导条件下为41%;乳酸/甲醇诱导条件下为39%。实验结果表明,在甲醇中混入不同的碳源均能减少酵母细胞的死亡,其中山梨醇效果最好,其次是乳酸>甘露醇>甘油。
2.4混合碳源的添加对胞内AOX酶活的影响
开始流加混合碳源7h时(也就是诱导10h),不同碳源的诱导下AOX的酶活分别是:仅甲醇诱导下酶活为1.2U/g;甘油/甲醇诱导下酶活为0.3U/g;山梨醇/甲醇诱导下酶活为0.6U/g;甘露醇/甲醇诱导下酶活为0.6U/g;乳酸/甲醇诱导下酶活为0.5U/g。诱导50h时,不同碳源的诱导下AOX的酶活分别是:甲醇诱导下酶活为3.9U/g;甘油/甲醇诱导下酶活为3.0U/g;山梨醇/甲醇诱导下酶活为3.7U/g;甘露醇/甲醇诱导下酶活为3.8U/g;乳酸/甲醇诱导下酶活为3.9U/g。诱导结束时,不同碳源的诱导AOX的酶活分别是:甲醇诱导下酶活为3.0U/g;甘油/甲醇诱导下酶活为5.0U/g;山梨醇/甲醇诱导下酶活为5.4U/g;甘露醇/甲醇诱导下酶活为5.7U/g;乳酸/甲醇诱导下酶活为5.5U/g。在流加甲醇的条件下,AOX酶活性在70h左右达到最大,为4.7U/g;流加甘油/甲醇的条件下,AOX酶活性在发酵结束时达到最大;流加山梨醇/甲醇,甘露醇/甲醇和乳酸/甲醇,AOX酶活性均在90h左右达到最大,分别为6.2、5.8、6.0U/g。
表8 流加不同的混合碳源对P.pastoris生长和h-LYZ的影响
实施例7诱导后期补加胰蛋白胨对h-LYZ的影响
在培养基中补加一些富含氨基酸和多肽的物质,为蛋白水解酶提供足量的底物,以减少目的蛋白的降解,进一步提高产物的稳定性。本实验在诱导36h后,每隔12h补加0.5%的胰蛋白胨,观察酶活和蛋白表达量。结果发现,补加胰蛋白胨后能够有效防止蛋白降解,提高酶活产量,在诱导96h后,酶活为53090U/mL,而对照组(不补加胰蛋白胨)在88h时酶活最高为47680U/mL。h-LYZ的活性能提高约10%,蛋白表达量无显著提高。具体结果如图18。
实施例8 50L和500L发酵罐放大及稳定性研究结果
在优化后的发酵条件下进行了50L和500L的工艺放大。在放大过程中发现,溶氧(DO)是影响菌体生长和h-LYZ表达的最重要因素,理论上来说,溶解氧速率应与菌体生长时耗氧速率相等,才能满足菌体对氧的需求。在实际操作中,结合了经验值与设备条件,在发酵过程中,根据DO的变化对空气通气量和搅拌速度进行相应的调整。整个过程的工艺是,在50L发酵罐中,初始装液量为20L,进罐接种量为5%,初始搅拌速度300rpm/min,初始通气量1.5vvm,罐压0.05MPa,菌体生长温度30℃,pH为5.5,DO控制在20%以上。当甘油耗尽时,DO迅速上升,然后以指数流加方式补加50%的甘油至一定菌浓要求。当再次甘油 耗尽,DO反弹约1h后,开始流加诱导培养基,温度降为24℃,pH为4.5,当诱导36h后,每隔12h补加0.5%的胰蛋白胨。500L发酵罐中,先是在1L摇瓶里进行一级种子培养,按照5%接种量至50L发酵罐中作为二级种子培养,然后按照10%接种量至500L发酵罐中。考虑生产成本,在进行500L放大时没有补加胰蛋白胨。具体的发酵结果和在线检测图见表9、图19和20。
50L放大结果:诱导90h后,菌体干重最高为180g/L,酶活最高为151600U/mL,蛋白表达量最高达1.65g/L,且批次具有较好的稳定性。
500L放大结果:诱导96h后,菌体干重为148g/L,酶活最高达127300U/mL,蛋白表达量约1.5g/L。500L放大结果理想,为工业化生产提供了实际参考依据。发酵过程中的溶氧、pH、尾碳、尾氧、转速等数据详见图20。
表9 50L发酵结果
SEQUENCE LISTING
<110> 复旦大学
<120> 一种人源溶菌酶蛋白发酵方法
<130> 201410
<160> 5
<170> PatentIn version 3.1
<210> 1
<211> 27
<212> DNA
<213> Artificial
<400> 1
gtgactcgag aaaagagagg ctgaagc 27
<210> 2
<211> 32
<212> DNA
<213> Artificial
<400> 2
taccgaattc ctagacaccg caaccttgga cg 32
<210> 3
<211> 21
<212> DNA
<213> Artificial
<400> 3
gactggttcc aattgacaag c 21
<210> 4
<211> 21
<212> DNA
<213> Artificial
<400> 4
gcaaatggca ttctgacatc c 21
<210> 5
<211> 453
<212> DNA
<213> Artificial
<400> 5
atgaaggctt tgattgtctt gggtctcgtt ttgctctccg tcactgttca gggtaaggtc 60
ttcgagagat gtgagttggc tagaaccttg aagagacttg gtatggacgg ttacagaggt 120
atttccttgg ctaattggat gtgcttggct aagtgggagt ctggctacaa taccagagct 180
actaactaca acgctggtga cagatccact gactacggta ttttccagat caattcccgt 240
tactggtgca acgacggtaa gaccccaggc gctgtcaatg cttgtcactt gtcctgttcc 300
gctttgctgc aagacaatat cgctgacgcc gtcgcctgtg ccaagagagt tgttagagac 360
ccacaaggta ttagagcttg ggtcgcttgg agaaatagat gtcaaaatag agacgttaga 420
caatacgtcc aaggttgcgg tgtctagtaa tga 453
- 还没有人留言评论。精彩留言会获得点赞!