新型基因打靶方法与流程
本发明涉及生物技术领域;具体地说,本发明涉及新型的打靶方法以及用于该方法的核苷酸构建物。
背景技术:
在生物体基因组中,各基因一般由编码区和调控区两部分组成。编码区或开放读框(ORF)编码具有各种生物学功能的蛋白和RNA链。蛋白编码序列的边界包括5’末端(N-末端)的起始密码子和3’末端(C-末端)的翻译终止无义密码子。编码区之前(5’区)和之后(3’区)的调控区含有各种DNA调控元件,例如启动子、增强子、终止子、多腺苷酸化信号,5'非翻译区(5'UTR)和3’非翻译区(3'UTR),它们控制基因表达过程的各个方面,包括转录、翻译和RNA稳定性等等。
生物基因表达主要有两个步骤:编码的基因从DNA转录成信使RNA(mRNA)或RNA,以及mRNA翻译为蛋白质。基因的表达分别可在转录和翻译步骤通过调控区各种DNA元件进行控制。
基因的转录通过启动子启动、延伸到终止子结束。启动子是基因序列中被RNA聚合酶识别并起始转录的特定区段,它是控制转录起始的序列并决定着基因的表达强度。终止子是基因序列中负责转录终止的特定序列,它提供信号触发转录机器释放新合成的mRNA(或RNA)终止转录。
翻译是以mRNA为模板合成蛋白质的过程。成熟的mRNA由三个部分组成:5'UTR、ORF和3'UTR。翻译起始复合物在5'至3'方向扫描5'UTR直至遇到起始密码子AUG,在该位置核糖体沿mRNA5’端向3’端移动,开始了从N端向C端的蛋白质合成。当核糖体遇到终止密码子(UAA、UAG或UGA)时,蛋白质合成终止,蛋白质从核糖体释放。
实际上,能可预测地调控任何目标基因表达的方法和/或工具将有益于生物学研究和众多生物技术应用。
基因打靶广泛用于破坏或增强染色体基因组中基因的活性。这是一种将外源DNA整合在遗传基因组中的方法,其导致靶基因被修饰、替换或复制。基因 打靶是对所有生命体通用的过程并可以用于任何基因,而且与基因的转录和翻译步骤无关。
基因打靶通常经由DNA双链断裂(DSB)的修复介导而实现。这种修复机制通过两种截然不同的分子机制发生:同源重组(HR)途径和非同源末端连接(NHEJ)途径。在同源重组基因打靶中,外源DNA片段(一般为可选择的标记基因)通过两端的同源序列精确整合在基因组的相应序列中。但是在非同源末端连接途径中,外源DNA片段会随机整合在非同源的染色体基因位点上(Paques and Haber 1999,Microbiology and Molecular Biology Reviews,63:349–404)。因此,位点特异性基因打靶的效率由同源重组和非同源末端连接途径之间的相对强度决定。
虽然非同源末端连接途径被认为是造成基因打靶效率低的主要原因,但在具有相同遗传背景的菌株中,同源重组基因打靶的效率也可能是基因依赖性的。这种基因依赖性现象的分子机制不十分清楚。一种可能的原因是各染色体基因组有热点区域,这些热点区域易于发生同源重组(Wahls et al.Plos One3:e2887)。
不同生物系统同源重组基因打靶的效率也大不相同。常规酵母,酿酒酵母(Saccharomyces cerevisiae)和裂殖酵母,粟酒裂殖酵母(Schizosaccharomyces pombe)已经有非常有效的同源重组基因打靶系统。然而,在甲醇营养型酵母,巴斯德毕赤酵母(Pichia pastoris)和其它“非常规”酵母,例如多形汉森酵母(Hansenula polymorpha)、脂耶罗威亚酵母(Yarrowia lipolytica)、树干毕赤酵母(Pichia stipitis)和乳酸克鲁维酵母(Kluyveromyces lactis)中,同源重组基因打靶效率极其低下。包括真菌和真核生物在内的大多数生物也具有极其低下的同源重组基因打靶效率(Klinner U,et al(2004)Fems Microbiology Reviews 28:201–223;Gregg JM(2010)Pichia Protocols,Second edition.Totowa,New Jersey:Humanna Press)。。
虽然基因打靶的效率取决于同源重组和非同源末端连接途径之间竞争、也依赖于基因组基因和生物系统,但对于基因的编码和调控区中不同位置的同源重组基因打靶效率知之甚少,特别是在难以破坏的基因中。
因此,本领域急需一种能提高基因打靶效率,特别是针对常规方法难以有效打靶的基因的基因打靶方法。
技术实现要素:
本发明的目的是提供一种能提高基因打靶效率,特别是针对常规方法难以有效打靶的基因的基因打靶方法以及用于这种方法中的物质手段。
在第一方面,本发明提供一种用于调控基因的核苷酸构建物,其结构如下式所示:
5’-A-B-C-3’
其中,A是5’同源序列,B是干扰基因,C是3’同源序列;
所述5’和3’同源序列使得所述核苷酸构建物的重组位点位于该待调控基因的起始密码子的第一个核苷酸到该待调控基因的起始密码子的第一个核苷酸上游的110,优选50个核苷酸之间,或者所述5’和3’同源序列使得所述核苷酸构建物的重组位点位于该待调控基因的终止密码子的第一个核苷酸上游的100、50或20个核苷酸,优选50个核苷酸到该待调控基因的终止密码子的第一个核苷酸下游的300个核苷酸。
在具体的实施方式中,所述重组位点之间间隔0-20个核苷酸,优选0-5个核苷酸,最优选0个核苷酸。
在优选的实施方式中,所述干扰基因可以多于一个,可以相同或不同。
在优选的实施方式中,所述干扰基因可以是标记基因。
在优选的实施方式中,所述核苷酸构建物可以是环状或线形的。
在具体的实施方式中,所述待调控基因可以是重组效率低的基因,优选重组效率<3%,更优选重组效率<1%。
在具体的实施方式中,所述待调控基因是OCH1、ADE1基因。
在优选的实施方式中,所述同源序列的长度为400-1200nt、500-1000nt、600-800nt。
在第二方面,本发明提供一种包含本发明第一方面所述的核苷酸构建物的宿主细胞。
在具体的实施方式中,所述宿主细胞是酵母细胞。
在优选的实施方式中,所述酵母是酿酒酵母(Saccharomyces cerevisiae)、粟酒裂殖酵母(Schizosaccharomyces pombe)、毕赤酵母(Pichia pastoris)、多形汉森酵母(Hansenula polymorpha)、脂耶罗威亚酵母(Yarrowia lipolytica)、树干毕赤酵母 (Pichia stipitis)和乳酸克鲁维酵母(Kluyveromyces lactis)。
在优选的实施方式中,所述酵母是毕赤酵母(Pichia pastoris)、多形汉森酵母(Hansenula polymorpha)、脂耶罗威亚酵母(Yarrowia lipolytica)、树干毕赤酵母(Pichia stipitis)和乳酸克鲁维酵母(Kluyveromyces lactis)。
在第三方面,本发明提供一种调控基因表达的方法,所述方法包括:
a)构建本发明第一方面所述的核苷酸构建物;和
b)将步骤a)构建的核苷酸构建物导入细胞,从而通过同源重组整合入待调控基因。
在具体的实施方式中,所述待调控基因可以是重组效率低的基因,优选重组效率<3%,更优选重组效率<1%。
在优选的实施方式中,所述待调控基因是OCH1、ADE1基因
在优选的实施方式中,所述方法还可包括步骤c)检测步骤b)所得细胞中待调控基因的表达。
在第四方面,本发明提供一种改造菌株的方法,包括:
a)构建本发明第一方面所述的核苷酸构建物;和
b)将步骤a)构建的核苷酸构建物转化入待改造的菌株。
在优选的实施方式中,所述方法还可包括步骤c)筛选改造菌株的步骤。
在第五方面,本发明提供一种采用本发明第四方面所述方法改造的菌株的用途,所述菌株用于生产重组蛋白。
在优选的实施方式中,所述重组蛋白中的糖基化模式得到改变。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
图1是典型的基因图谱,包括5’调控区(5’区)、开放读框(ORF)和3’调控区 (3’区)。启动子和增强子决定该基因的哪部分被转录为信使RNA(mRNA)。5’和3’UTR调控mRAN到蛋白质的翻译过程。5'和3'区的核苷酸编号是指编码区的相应起始密码子作为核苷酸1-3,其5’上游区域用减号编号;而相应终止密码子作为核苷酸+1至+3,其3'下游区域用加号编号。载体组分未按比例绘制。
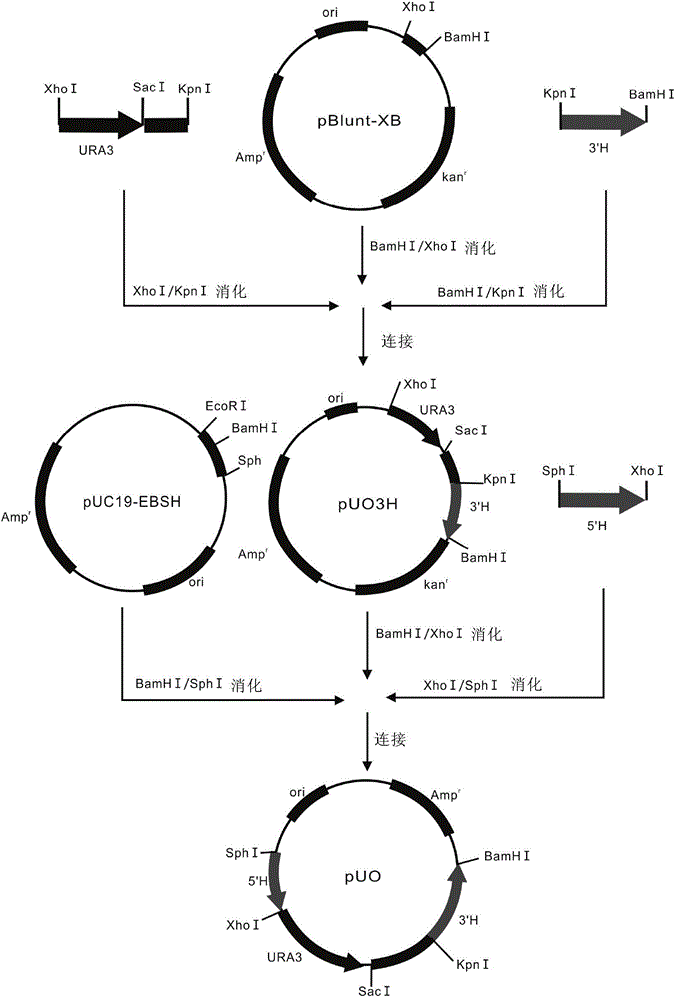
图2描述了构建pUO载体的示意图。载体组分未按比例绘制。
图3描述了构建pUAH(1)打靶载体以便整合入毕赤酵母的OCH1基因的示意图。载体组分未按比例绘制。
图4描述了在毕赤酵母的OCH1基因(SEQ ID NO:126)中不同位置整合的示意图。载体组分未按比例绘制。A.毕赤酵母OCH1基因座中打靶盒的整合位置。OCH1基因座的整合位置由箭头表示并标有核苷酸编号。载体中的基因组分未按比例绘制。B.描述了打靶盒通过双交换同源重组整合入毕赤酵母的OCH1基因(敲入)。交叉表示同源重组。C.显示OCH1基因中不同位置处整合的PCR验证结果。M,DNA大小标记物;泳道1,用P1/P4引物对,JC301中的野生型OCH1得到1433bp;泳道2、3、4,3个位置(-1/1)、(1212/+1)和(+3/+4)的基因整合未得到条带,因为序列太长(4900bp),PCR用P1/P4引物对无法扩增;泳道5,用P1/P2引物对,JC301中的野生型OCH1未得到条带;泳道6、7、8,用P1/P2引物对,3个位置(-1/1)、(1212/+1)和(+3/+4)的基因整合分别得到1300、2550和2550bp带;泳道9,用P3/P4引物对,JC301中的野生型OCH1未得到条带;泳道10、11、12,用P3/P4引物对,3个位置(-1/1)、(1212/+1)和(+3/+4)的基因整合分别得到3500、2500和2300bp条带。
图5描述了在毕赤酵母基因组的ADE1基因(巴斯德毕赤酵母PR-氨基咪唑琥珀酰羧酰胺合酶(ADE1)的DNA序列如SEQ ID NO:127所示)中不同位置整合的示意图。载体组分未按比例绘制。A.毕赤酵母ADE1基因中打靶盒的整合位置。整合位置由箭头表示并标有核苷酸编号。B.描述了打靶盒通过双交换同源重组整合入毕赤酵母的ADE1基因座(敲入)。交叉表示同源重组。C.显示ADE1基因中不同位置处整合的PCR验证结果。M,DNA大小标记物;泳道1-13,用P5/P6引物对,通过PCR验证13个随机选择菌落的基因组DNA。野生型ADE1得到2398bp条带,位置(912/+1)的基因整合得到3763bp条带。
图6描述了构建5’AOX1诱导启动的lacZ表达载体p5’AOX1-URA3-lacZ的示意图,其中URA3位于5’AOX1和lacZ ORF之间。载体组分未按比例绘制。
图7描述了构建一系列5’AOX1诱导启动的lacZ和lacZns表达载体p5’AOX1-lacZ-URA3、p5’AOX1-lacZ-URA3(-)、p5’AOX1-lacZns-URA3和 p5’AOX1-lacZns-URA3(-)的示意图,其中URA3以两种取向位于lacZ和lacZns下游。载体组分未按比例绘制。
图8描述了构建一系列5’OCH1启动的lacZ和lacZns表达载体p5’OCH1-lacZ和p5’OCH1-lacZns的示意图。载体组分未按比例绘制。
图9描述了构建5’OCH1启动的lacZ表达载体p5’OCH1-URA3-lacZ的示意图,其中URA3位于5’OCH1和lacZ ORF之间。载体组分未按比例绘制。
图10描述了构建一系列5’OCH1启动的lacZ表达载体p5’OCH1-lacZ-URA3和p5’OCH1-lacZ-URA3(-)的示意图,其中URA3以两种取向位于lacZ的下游。载体组分未按比例绘制。
图11描述了构建一系列5’OCH1启动的lacZns表达载体p5’OCH1-lacZns-URA3和p5’OCH1-lacZns-URA3(-)的示意图,其中URA3以两种取向位于lacZns的下游。载体组分未按比例绘制。
图12显示了lacZ mRNA的相对表达量(%)。A.在毗邻起始和终止密码子的URA3存在下,5’AOX1诱导启动的lacZ mRNA相对表达量(%)。100%对应于无URA3整合下(p5’AOX1-lacZ),5’AOX1诱导启动的lacZ mRNA表达量。B.在毗邻起始和终止密码子的URA3存在下,5’OCH1启动的lacZ mRNA相对表达量(%)。100%对应于无URA3整合下(p5’OCH1-lacZ),5’OCH1启动的lacZ mRNA表达。数据显示为3次实验的平均值±标准偏差(s.d.)。
图13显示了β-半乳糖苷酶在细胞内的相对活性(%)。A.在毗邻起始和终止密码子的URA3存在下,5’AOX1诱导启动的β-半乳糖苷酶活性。100%对应于无URA3整合下(p5’AOX1-lacZ),5’AOX1诱导启动的β-半乳糖苷酶活性。B.在毗邻起始和终止密码子的URA3存在下,5’OCH1启动的β-半乳糖苷酶活性。100%对应于无URA3整合下(p5’OCH1-lacZ),5’OCH1启动的β-半乳糖苷酶活性。数据显示为3次实验的平均值±标准偏差(s.d.)。
图14显示了在毗邻起始和终止密码子含有基因整合的菌株中OCH1 mRNA的相对表达量(%)。100%对应于无基因整合的亲代JC307菌株中的mRNA表达。数据显示为3次实验的平均值±标准偏差(s.d.)。
图15显示mIL-22释放的N-糖链的正离子MALDI-TOF质谱图。A.显示用GS115菌株表达的mIL-22释放的N-糖链的质谱图。B.显示用菌株och1(-1/+1,ADE1URA3)表达的mIL-22释放的N-糖链的质谱图,该菌株在OCH1ORF上游有基因整合。
具体实施方式
发明人经过广泛而深入的研究,出乎意料地发现了不依赖于基因的同源重组有效基因打靶区域,基于此还开发了用于基因表达调控和基因破坏的基因打靶系统,本发明方法可调控或修饰生物体的任何基因。在此基础上完成了本发明。
本发明系统地分析基因中不同位置的基因打靶,鉴定了基因中同源重组的有效整合区。还开发了基因打靶系统以供基因表达调控和基因破坏。本发明利用所有生物体细胞中内源性的同源重组过程,因此,本发明方法可调控或修饰生物体的任何基因。本发明方法在生物科技产业和生物学研究领域可广泛用于调控基因表达、改进细胞功能和生产异源蛋白质。
以下术语根据下面的定义在本文使用。
“基因”作广义使用,指生物学功能相关的任何核酸区段。
“肽”、“多肽”和“蛋白质”在本文可互换使用,是指任何长度的聚合形式氨基酸。
"基因打靶"是在遗传基因处整合外源DNA的方法,通常导致在靶基因处修饰、替换或复制。这是对所有生命体通用的机制。
"细胞"或"机体"是用于实施本发明的基因打靶的机体的术语。
"细胞转化"表示外源DNA被引入细胞。这通常是外源DNA整合入染色体DNA或引入自我复制质粒所致。
"靶基因"或"靶部位"是指通过本发明的基因打靶方法予以改变的基因或DNA区段。靶基因可以是内源基因或先前引入机体的外源DNA区段。靶基因可以是机体的内源性基因组DNA、基因的任何部分,包括但不限于多肽编码区、开放读框、调控区、内含子、外显子或它们的一部分。
"标记"代表基因序列,其存在或不存在提供机体的可检测表型。不同类型的标记包括但不限于:选择标记、筛选标记和分子标记。选择标记通常是基因,其表达可以使得机体具有对一组特定条件耐受或敏感的表型。筛选标记传递作为可观察和可区分特征的表型。分子标记是可由DNA分析鉴定的基因序列特征。
基因包括编码特定蛋白或功能性RNA的“编码序列”、“编码区”或“开放读框”。蛋白编码序列是转录为mRNA的核酸序列,进而翻译成蛋白质。蛋白编码序列的边界由5’末端(N-末端)的起始密码子和3’末端(C-末端)的翻译终止无义密码子界定。
基因还包括在编码序列之前和之后的“调控区”或“调控元件”。所述调控元件包括但不限于启动子、增强子、内含子、多腺苷酸化信号,5'非翻译区(5'UTR)、3’非翻译区(3'UTR)和它们的任何衍生物。一些调控区转录为RNA分子的一部分,例如5'UTR和3'UTR。术语“5’非翻译区(5′UTR)”应表示成熟mRNA中紧邻任何编码序列上游的核苷酸序列,其不翻译成蛋白质。术语“3’非翻译区(3′UTR)”应表示成熟mRNA中紧邻任何编码序列下游的核苷酸序列,其不翻译成蛋白质。其从任何编码序列的终止密码子之后的第一个核苷酸延伸恰到mRNA的poly(A)尾之前。这些调控元件可以调控基因表达过程的各个方面,包括但不限于转录(例如,启动、延伸和/或终止)、翻译(启动、延伸和/或终止)和RNA稳定性,等等。
“启动子”是能聚合RNA聚合酶并启动下游(3’方向)编码序列转录的核酸调控区。启动子序列在其3’末端以转录起始位点为界,向上游延伸(5’方向),包括启动转录所需的最低数量的碱基或元件。启动子序列中可以找到转录起始位点以及负责结合RNA聚合酶的蛋白质结合域。真核启动子常(但不总是)含有“TATA”盒和“CAT”盒,而原核启动子常含有共有序列TATAAT。许多启动子称为组成型启动子,因为它们在细胞的所有环境中具有活性,但一些是诱导型启动子,其活性通过对特定刺激起反应而受到调控。
“终止子”是核酸序列的某区段,其在转录期间提供信号触发转录机器释放新合成的mRNA(或RNA)终止转录。在原核转录中,两类转录终止子,依赖于Rho和不依赖于Rho的序列负责触发转录终止。在真核mRNA转录中,转录机器识别终止子信号并触发终止过程释放mRNA,随后poly(A)序列通过多腺苷酸化反应加在mRNA的3'端。
对转录的调控根据其作用性质主要可以分为两大类:第一类是DNA模板功能抑止,通过抑止分子与DNA结合改变模板功能;第二类是RNA聚合酶抑止,通过抑止分子与RNA聚合酶结合而抑止其活性(Sandhya Payankaulam,Li M.Li,and David N.Arnosti(2010)Transcriptional repression:conserved and evolved features.Curr Biol.14;20(17):R764–R771)。然而,这两类控制方法不能用作通用方法来专一性地调节任何靶基因的表达。
“翻译”是以mRNA为模板合成蛋白质的过程。成熟的mRNA由三个部分组成:5'UTR、ORF和3'UTR。翻译起始复合物在5'至3'方向扫描5'UTR直至遇到起始密码子AUG,在该位置核糖体沿mRNA5’端向3’端移动,开始了从N端向C端的蛋白质合成。当核糖体遇到终止密码子(UAA、UAG或UGA)时,蛋白质合成终止,蛋白质从核糖体释放。
翻译过程可由RNA-结合蛋白(RBP)和小RNA控制,它们通过结合在mRNA上来调控其翻译。RBP通常结合在位于5’或3’UTR中的特定元件以激活或阻遏翻译。然而,5’UTR内的元件处于扫描/翻译核糖体的途径内,核糖体能在调控元件发生作用前置换它们。通过检测细胞内所有mRNA的降解率和翻译率时发现,对两者影响最高的3’UTR元件是microRNA互补位点,它对mRNA的稳定性和翻译水平分别抑制32%和4%。然而,3’UTR对大多数mRNA的稳定性和翻译效率的影响有限(Noah Spies,Christopher B.Burge,and David P.Bartel(2013),3’UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts,Genome Research,23:2078–2090)。因此,通过RBP和小RNA结合在mRNA上来调节蛋白质翻译应该不会是一种有效的方法。
细胞具有监督系统来识别和消除异常mRNA以避免产生可能有害的蛋白质产物。例如,细胞能识别缺少终止密码子的异常mRNA(无终止mRNA)并在3’端形成Ski复合物以介导该无终止mRNA的降解。这种无终止mRNA衰减可避免产生可能有害的延伸产物,这种产物相对于野生型基因产物具有显性负活性(van Hoof A,Frischmeyer PA,Dietz HC,Parker R(2002)Exosome mediated recognition and degradation of mRNAs lacking a termination codon.Science 295:2262–2264)。
实际上,目前缺少调控基因组基因表达的有效方法。任何能够可预测专一性地调控基因组中目标基因表达的方法和/或工具都将有益于生物学研究和生物技术发展。
基因打靶技术是一种将外源DNA整合在染色体遗传基因组中的方法,从而 导致靶基因处的基因被修饰、替换或复制,它广泛用于破坏基因活性。
“Ends-in”和“Ends-out”是指可用于经同源重组整合外源DNA进入基因组的两种不同设置。在通过“Ends-in”重组的基因打靶中,当外源DNA与基因组中的同源区域配对时,线形外源DNA的末端指向彼此,通过单交换重组(single cross-over recombination,“roll in”)将DNA整合进入基因组,产生靶基因同向重复序列。但是重复的靶基因可以再通过自身的同源重组切除外源DNA,恢复靶基因原来的野生型状态。在通过“ends-out”重组的基因打靶中,当外源DNA与基因组中的同源区域配对时,线形外源DNA的末端背离彼此,通过末端靶向侧翼和宿主基因组同源序列之间的双交换重组将DNA插入基因组中。“Ends-out”打靶常用于小鼠和酵母,因为它可以直接替换或删除靶基因。不过,“ends-out”事件发生的概率远低于“ends-in”事件。(Paques and Haber 1999,Microbiology and Molecular Biology Reviews,63:349–404)。在本发明中,基因打靶是指“ends-out”双交换重组,除非专门表明是通过单交换的“ends-in”打靶(roll-in)。
基因打靶是对所有生命体通用的过程并可以用于任何基因,而且不受其转录和翻译活性影响。然而,该技术受限于以下两个局限性:同源重组效率低和随机(非靶向)整合率高。
基因打靶藉由两种截然不同的分子机制发生:同源重组(HR)途径和非同源末端连接(NHEJ)途径。两种重组途径通常经由DNA断裂双链(DSB)的修复介导。在同源重组基因打靶中,外源DNA片段,通常是两端有同源序列的可选择标记基因,精确整合在其同源基因组的相应序列中。但是在非同源末端连接途径中,含可选择标记基因的外源DNA片段会随机整合在非同源的染色体位点上。位点特异性基因打靶的效率通常由同源重组和非同源末端连接途径之间的相对强度决定。
不同生物系统同源重组基因打靶的效率也大不相同。常规酵母,酿酒酵母(Saccharomyces cerevisiae)和裂殖酵母,粟酒裂殖酵母(Schizosaccharomyces pombe)具有非常有效的同源重组基因打靶系统。在酿酒酵母中,当打靶片段是30至45bp时,同源重组基因打靶替换的效率最高可达到转化菌的95%(Paques and Haber 1999,Microbiology and Molecular Biology Reviews,63:349–404)。然而,大多数生物具有极低的同源重组基因打靶效率。在甲醇营养型酵母,巴斯 德毕赤酵母(Pichia pastoris)和其它“非常规”酵母,包括多形汉森酵母(Hansenula polymorpha)、脂耶罗威亚酵母(Yarrowia lipolytica)、树干毕赤酵母(Pichia stipitis)和乳酸克鲁维酵母(Kluyveromyces lactis)中,基因打靶效率极低。基因打靶替换的效率高度依赖于打靶片段中同源序列的长度。当打靶同源序列小于500bp时,效率可能低于0.1%,但是当利用大的1kb的打靶同源序列时,对于一些靶基因可以高于50%(Klinner U,et al(2004)Fems Microbiology Reviews 28:201–223;Gregg JM(2010)Pichia Protocols,Second edition.Totowa,New Jersey:Humanna Press)。包括真菌和真核生物在内的大多数生物同源重组基因打靶效率也很低。
此外,在具有相同遗传背景的菌株中同源基因打靶的效率还是基因依赖性的。例如,当打靶同源序列的长度在200-900bp时,毕赤酵母GS115菌株中的ARG1、ARG2、ARG3、HIS1、HIS2、HIS5和HIS6以44-90%的高效率发生同源重组(Nett,et al(2005)Yeast 22:295–304)。但当利用约1kb或更长的同源序列去敲除毕赤酵母基因组中OCH1和SGS1基因时,其效率低于1%(Choi,et al.(2003)Proc Natl Acad Sci U S A 100:5022–5027;Chen,et al.(2013)PLoS ONE8(3):e57952)。这种基因依赖性现象的分子机制不十分清楚。一种可能的原因是各染色体有热点区域,这些热点区域易于发生同源重组(Wahls et al.Plos One 3:e2887)。
虽然基因打靶的效率取决于同源重组和非同源末端连接途径之间竞争、也依赖于基因组基因和生物系统,但对于基因的编码和调控区中不同位置的同源重组基因打靶效率知之甚少,特别是在难以破坏的基因中。
本发明利用OCH1,一种低效的打靶基因,和ADE1,一种常见的打靶基因,作为模型来系统性分析基因组基因中各种位置的同源基因打靶。本项研究有助于将基因打靶技术体系应用于基因表达调控和基因破坏。
本发明开发了打靶载体,包括标记基因、同源序列和复制起点的诸部分。这些部分可以相连形成环状载体。如果需要的话,该环状载体在诸部分之间可以含有其它部分和接头。然而,本发明也应包括功能等价的其它形式打靶载体。打靶载体也可称为载体。用于重组DNA技术的载体通常是"质粒"的形式。在本说明书中,术语"载体"和"质粒"可互换使用。
"标记"代表基因或序列,其存在或不存在提供机体的可检测表型。可利用 一种或多种标记选择和筛选基因打靶事件。可用于本发明的不同类型的标记包括但不限于:选择标记、筛选标记和分子标记。
选择标记基因的表达可以使得机体具有对一组特定条件耐受或敏感的表型。选择标记包括对抗生素,例如卡那霉素、潮霉素、zeocin、博莱霉素、壮观霉素、链霉素、庆大霉素等耐受的基因。
可选择标记体系由营养缺陷突变型宿主菌株和野生型生物基因构成,补充宿主对不完全培养基的缺陷,例如酵母菌中的HIS4、LEU2、URA3、ADE1、LYS2和TRP1基因,和本领域已知的其它基因。例如,酿酒酵母或毕赤酵母HIS4基因可用于his4毕赤酵母菌株的转化。
筛选标记传递可观察和可区分的特征。可筛选标记包括荧光蛋白,例如绿色荧光蛋白(GFP)、报道酶,例如β-半乳糖苷酶(lacZ)、碱性磷酸酶(AP)、β-内酰胺酶、β-葡萄糖醛酸酶、谷胱甘肽S-转移酶(GST)、荧光素酶和本领域已知的其它酶。
分子标记是可由DNA分析鉴定的基因序列特征。
标记基因侧接两个同源重组区。所述同源重组区之一的上游侧与靶基因的上游区域同源,所述同源重组区之一的下游侧与靶基因的下游区域同源。在上、下游同源重组区之间单个或多个标记基因相互间可以以相同或相对的取向相连结。
所述同源重组区使得重组位点在待调控基因的起始密码子的第一个核苷酸到该待调控基因的起始密码子的第一个核苷酸上游的110、优选50个核苷酸之间,或者所述5’和3’同源序列使得所述核苷酸构建物的重组位点位于待调控基因的终止密码子的第一个核苷酸上游的100、50或20个核苷酸,优选50个核苷酸到该待调控基因的终止密码子的第一个核苷酸下游的300个核苷酸。
在本文,某区域与相应基因区域同源表示该区域与所述基因区域的碱基序列有至少90%、优选至少92%、更优选至少94%、还要更优选至少96%、还要更优选至少98%、还要更优选至少99%、最优选100%相同。优先的选择是这种“同源区域”源自所述的基因区域。
同源重组区域的长度不作特别限定。该区域的长度优选适于发生同源重组。因此,该区域的长度至少40个碱基对。
当考虑将本发明的载体转入细菌细胞传代时,优选载体中包含细菌复制起点和抗生素耐受性基因,以确保细菌传代过程中保有该载体。细菌复制起点(ori)包括fl-ori、colisin、col El和本领域已知的其它起点。抗生素耐受性基因包括氨苄青霉素、卡那霉素、四环素、Zeocin耐受性基因和本领域已知的其它抗生素耐受性基因。复制起点和抗生素耐受性基因可以连接在不同部分之间。
本发明提供线形“打靶盒”,可利用限制性酶消化使之从打靶载体线形化,或者可通过基因化学合成得到。为简便起见,该“打靶盒”在本文还可称为“打靶片段”、“基因破坏片段”、或“基因整合的片段”。该打靶盒用于破坏靶基因并将外源基因整合入宿主的染色体基因组中,从而该外源基因能在该宿主中行使功能。
打靶盒的实质性部分包括标记基因和同源区。打靶盒可含有其它部分,如果需要,可在诸部分之间含有接头。标记基因的上游和下游侧侧接同源区域。
将打靶盒或载体引入宿主细胞以供同源重组。可按照本领域技术人员熟知的方法进行宿主细胞的转化和转染。
合适的转化方法包括病毒感染、转染、接合、原生质体融合、电穿孔、基因枪技术、磷酸钙沉淀、直接显微注射等等。方法的选择通常依赖于所转化的细胞类型和进行转化的条件。这些方法的常规讨论见Ausubel,et al.,Short Protocols in Molecular Biology,3rd ed.,Wiley & Sons,1995。
例如,可采用不同的方法实施酵母转化,包括球形体方法、电穿孔、聚乙二醇方法、碱性阳离子方法等等[Gregg JM(2010)Pichia Protocols,Second edition.Totowa,New Jersey:Humanna Press]。
可用于本发明的宿主细胞的例子包括典型的真核的和原核宿主,例如大肠杆菌(E.coli)、假单胞菌属(Pseudomonas spp.)、芽胞杆菌属(Bacillus spp.)、链霉菌属(Streptomyces spp.)、真菌、酵母菌,例如酿酒酵母、毕赤酵母,昆虫细胞,例如草地贪夜蛾(Spodoptera frugiperda)(SF9),动物细胞,例如中国仓鼠卵巢细胞(Chinese hamster ovary cell)(CHO)和小鼠细胞,非洲绿猴细胞,培养的人类细胞和植物细胞。酵母是优选的本发明宿主细胞。毕赤酵母是更优选的宿主细胞。
然后可基于可选标记的表型选择标记选择转化的细胞。
本发明评估了5’-调控区、编码区和3’-调控区的位点特异性同源重组整合,鉴定了高频率基因整合的区域。以前的报道说,当利用1kb或更大同源区域时,同源重组基因打靶的效率依赖于基因组基因,与之相反,本发明发现,当利用小于1kb同源(区域)时,5’-和3’-调控区的同源重组基因整合效率不依赖于基因,其水平显著较高。此外,编码区3’端基因整合的频率高于其它编码区。基因组基因的不同区域中打靶整合的频率可以表示为以下次序:5’-调控区和3’-调控区﹥﹥编码区的3’端﹥其它编码区。
本发明开发了通过在5’-调控区作基因整合来精确控制靶基因表达的方法。整合基因可以是有助于鉴定具有基因整合的转化株的任何标记基因,包括可选择标记体系、筛选标记和分子标记。标记基因ORF可与不同启动子、分泌信号序列(如果需要的话)和转录终止子按一定的位置和方向排列,融合形成表达盒。这些区段的排列位置和方向是本领域普通技术人员已知的。这些标记基因表达盒可以在基因组的DNA双链中,与靶ORF以相同或相对的方向整合在5’-调控区的任何位置,更优选5’-调控区中接近靶ORF的位置,最优选在同一链中,相同取向的紧邻靶ORF上游的位置。也可以将标记基因ORF和转录终止子融合体,整合在5’-调控区同一链中,相同取向接近靶ORF的位置,最优选整合在紧邻靶ORF上游的位置,如此可利用靶基因的5’-调控区以启动标记基因的表达。
5’调控区特别是在紧邻靶ORF上游位置的基因整合可以抑制转录和翻译的效率,从而有效地抑制特定靶基因表达。目前调控基因转录的方法主要包括:通过抑止分子与DNA结合改变模扳功能,通过抑止分子与RNA聚合酶结合而抑止其转录活性。调控蛋白翻译的方法主要采用小RNA和RNA-结合蛋白与mRNA结合来改变其翻译能力。相比之下,在基因组基因中,特别是在ORF上游进行的基因整合是更有效的精确控制靶基因表达的方法。
在另一方面,可通过在接近靶ORF起始密码子的5’区中的任何位置,更优选起始密码子上游,最优选起始密码子上游3-10个碱基的位置整合打靶盒引进强启动子来上调任何靶ORF表达,所述打靶盒由在下游与强启动子融合的标记基因表达盒构成。
另一方面,本发明开发了通过在基因组的DNA双链中,以相同或相对取向在3’-调控区,最优选在紧邻终止密码子下游的位置整合选择标记盒来降低靶 ORF基因表达的方法。
另一方面,本发明开发了通过在基因组的DNA双链中,以相同或相对取向在编码区3’端附近整合选择标记盒来降低靶ORF基因表达的方法。
下表1比较了毕赤酵母的OCH1和ADE1基因中不同位置的同源重组整合频率。这些位置由基因组基因的核苷酸编号确定,所述编号是指编码区的相应起始密码子作为核苷酸1-3和相应终止密码子作为核苷酸+1至+3。正确的整合子是通过PCR验证的具有正确基因整合的克隆。打靶频率定义为通过PCR验证的正确整合子与检查的总克隆之比。
表1
基于本发明的基因打靶的方法,技术人员能够改造菌株,具体地说,能够改造菌株中利用常规打靶方法同源重组效率低于3%,优选低于1%的基因。进一步地,本发明提供了改造菌株的方法,这种改造的的菌株可用于生产重组蛋白。在具体的实施方式中,所述重组蛋白中的糖基化模式得到改变。例如,通过破坏OCH1基因,使改造的菌株不会对表达的蛋白过度糖基化修饰而产生不同于哺乳动物细胞的高甘露糖链Man9-100GlcNAc2;通过破坏菌株中的蛋白酶基因,可以减少重组蛋白的降解;等等。通过本发明方法改造的菌株还可用于代谢工程改造、遗传学研究以及生物技术应用等各个领域。
本发明的优点:
1.本发明鉴定到不依赖于基因的有效同源重组基因打靶的区域;
2.本发明方法可调控或修饰生物体的任何基因;
3.本发明方法在生物科技产业和生物学研究领域可广泛用于调控基因表达、改进细胞功能和生产异源蛋白质。
实施例
材料
用于文库的产生、验证和应用的化学试剂、酶、培养基和溶液是常用的并且是分子和细胞生物学领域的技术人员熟知的;它们可以从许多公司获得,包括Thermo Fisher Scientific、Invitrogen、Sigma、New England BioLabs、Takara Biotechnology、Toyobo、TransGen Biotech和Generay Biotechnology等等。其中许多以试剂盒的形式提供。
pPIC3.5K和pPICZ载体获自Invitrogen。
pBLHIS-SX、pBLURA-SX、pBLADE-SX载体获自Keck Graduate Institute,Claremont,加拿大。
大肠杆菌(E.coli)菌株Trans1-T1获自TransGen Biotech。
毕赤酵母营养缺陷型菌株JC301(ade1 his4 ura3)和JC307(his4 ura3)获自Keck Graduate Institute(KGI),GS 115(his)获自Invitrogen。
核苷酸序列数据主要获自公共数据库NCBI(www.ncbi.nlm.nih.gov)。
方法
除非另有表示,按照分子和细胞生物学领域技术人员熟知的标准方法进行本发明所用的方法,包括聚合酶链式反应(PCR)、限制性酶克隆、DNA纯化、细菌和原核细胞培养、转化、转染和蛋白质印迹,例如以下手册所述的:Sambrook J et al.(Molecular Cloning A Laboratory Manual(Third Edition),Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.,2001),Ausubel F M et al.(Current Protocols in Molecular Biology,Wiley InterScience,2010),and Gregg JM(Pichia Protocols,(Second edition),Totowa,New Jersey:Humanna Press,2010)。
大肠杆菌菌株Trans1-T1用于构建和扩增质粒。菌株用含合适抗生素的Luria-Bertani(LB)培养基(10g/L胰蛋白胨、5g/L酵母提取物和5g/L氯化钠)或LB平板(15g/L琼脂)培养。抗生素的加入浓度如下所述:100mg/L氨苄青霉素、50mg/L卡那霉素和25mg/L Zeocin)。
毕赤酵母菌株利用YPD培养基(10g/L酵母提取物、20g/L蛋白胨、20g/L葡萄糖)和YPD平板(10g/L酵母提取物、20g/L蛋白胨、20g/L葡萄糖、15g/L琼脂)培养。利用不含氨基酸的YNB培养基(67g/L酵母氮源(yeast nitrogen base)、5g/L葡萄糖)和不含氨基酸的YNB平板(67g/L酵母氮源、5g/L葡萄糖,15/L琼脂)来选择毕赤酵母营养缺陷型菌株,视需要适当添加(抗生素)。一些毕赤酵母营养缺陷型菌株利用SC培养基(8g/L SC不含组氨酸和尿嘧啶,20g/L葡萄糖)和SC平板(8g/L SC不含组氨酸和尿嘧啶,20g/L葡萄糖,15/L琼脂)选择,视需要适当添加(抗生素)。抗生素的加入浓度如下所述:500mg/L G-418硫酸盐和100mg/L Zeocin。
采用乙酸锂-SDS裂解,然后进行乙醇沉淀提取毕赤酵母中的基因组DNA,该方法描述于以下出版物:Looke et al.2011,Biotechniques.50:325–328。
利用MicroPulserTM电穿孔设备,按照生产商(BioRad)的操作使用说明书,通过电穿孔进行毕赤酵母的转化。
实施例1
构建基础载体
图2描述了构建pUO载体的示意图。
PCR1,用基因组DNA作为模板,用KpnIOch1(+54)F(SEQ ID NO:1,该引物具有Kpn I限制性酶切位点)和Och1(+801)BamHI R(SEQ ID NO:2,该引物具有BamH I限制性酶切位点)引物对作PCR扩增毕赤酵母OCH1 3’序列(3’H);
PCR2,利用pBlunt-URA3SK载体作为模板,利用XhoIURA3 F(SEQ ID NO:3,该引物具有Xho I限制性酶切位点)和DRKpnI R(SEQ ID NO:4,该引物具有Kpn I限制性酶切位点)引物对作PCR扩增毕赤酵母URA3表达盒和SacI-KpnI片段。通过将URA3和SacI-KpnI片段的PCR融合片段连接于pBlunt载体(TransGen Biotech,中国)获得pBlunt-URA3SK载体。
然后,分别用Kpn I和BamH I消化OCH1 3’H的PCR产物,用Xho I和Kpn I消化URA3表达盒。将OCH1 3’H的KpnI-BamHI片段和URA3表达盒的XhoI-KpnI片段插入pBlunt-XB载体的Xho I和BamH I位点以产生pUO3H载体。将含XhoI和BamHI位点的片段连接于pBlunt载体(TransGen Biotech)获得pBlunt-XB。
PCR3,用基因组DNA作为模板,利用SphIOch1(274)F(SEQ ID NO:5,该引物具有Sph I限制性酶切位点)和Och1(+53)XhoI R(SEQ ID NO:6,该引物具有Xho I限制性酶切位点)引物对作PCR扩增毕赤酵母OCH1 5’序列(5’H);
然后,分别用Sph I和Xho I消化OCH1 5’H的PCR产物,用Xho I和BamH I消化pUO3H载体。将OCH1 5’H的SphI-XhoI片段和URA3表达盒与OCH1 3’H相连的XhoI-BamHI片段插入pUC19-EBSH载体的BamH I和Sph I位点。用含EcoR I、BamH I、Sph I和Hind III限制性酶切位点的片段替换有多克隆位点的pUC19EcoRI-HindIII片段得到pUC19-EBSH载体。产生的pUO载体用作基础载体以构建其它不同的OCH1打靶载体。
实施例2
构建OCH1打靶载体
图3描述了构建整合入毕赤酵母OCH1基因的打靶载体的示意图。
PCR4,用毕赤酵母基因组DNA作为模板,用SacIADE1 F(SEQ ID NO:7,该引物具有Sac I限制性酶切位点)和ADE1KpnI R(SEQ ID NO:8,该引物具有Kpn I限制性酶切位点)引物对扩增ADE1表达盒。
PCR5,用pBLURA-SX(Keck Graduate Institute)作为模板,用SacIURA3F(SEQ ID NO:9,该引物具有Sac I限制性酶切位点)和URA3XhoI R(SEQ ID NO:10,该引物具有Xho I限制性酶切位点)引物对扩增毕赤酵母URA3表达盒。
然后,分别用Sac I和Kpn I消化ADE1表达盒的PCR产物,用Sac I和Xho I消化URA3表达盒的PCR产物。将ADE1的SacI-KpnI片段和URA3的SacI-XhoI片段插入pUO载体的Xho I和KpnI位点以产生pUAH载体。
PCR6,用基因组DNA作为模板,用SphIOch1(-733)F(SEQ ID NO:11,该引物具有Sph I限制性酶切位点)和Och1(-1)XhoI R(SEQ ID NO:12,该引物具有Xho I限制性酶切位点)引物对作PCR扩增毕赤酵母OCH1 5’同源序列(5’H, -733/-1)。
用限制性酶消化PCR产物后,将OCH1 5’H的SphI-XhoI片段(-733/-1)插入pUAH的相同限制性酶位点以产生pUA5H。
PCR7,用基因组DNA作为模板,用KpnIOch1(1)F(SEQ ID NO:13,该引物具有Kpn I限制性酶切位点)和Och1(646)BamHI R(SEQ ID NO:14,该引物具有BamH I限制性酶切位点)引物对作PCR扩增毕赤酵母OCH1 3’同源序列(3’H,1/646)。
用限制性酶消化PCR产物后,将OCH1 3’H的KpnI-BamHI片段(1/646)插入pUA5H的相同限制性酶位点以产生OCH1打靶载体pUAH(1),其用于整合入OCH1 5’调控区中紧邻起始密码子上游的位置(-1/1)。
以相同的方式,通过将OCH1 5’和3’同源性(序列)的相应PCR产物插入pUAH来构建一系列OCH1打靶载体,它们整合在OCH1基因的不同位置。
引物对SphIOch1(-118)F(SEQ ID NO:15)和Och1(553)XhoI R(SEQ ID NO:16)以及引物对KpnIOch1(554)F(SEQ ID NO:17)和Och1(+103)BamHI R(SEQ ID NO:18)用于构建pUAH(554)以便整合在OCH1编码区的中的位置(553/554)。
引物对SphIOch1(274)F(SEQ ID NO:19)和Och1(1096)XhoI R(SEQ ID NO:20)以及引物对KpnIOch1(1097)F(SEQ ID NO:21)和Och1(+801)BamHI R(SEQ ID NO:22)用于构建pUAH(1097)以便整合在OCH1编码区中的位置(1096/1097)。
引物对SphIOch1(274)F(SEQ ID NO:19)和Och1(1165)XhoI R(SEQ ID NO:23)以及引物对KpnIOch1(1166)F(SEQ ID NO:24)和Och1(+801)BamHI R(SEQ ID NO:22)用于构建pUAH(1166)以便整合在OCH1编码区中的位置(1165/1166)。
引物对SphIOch1(274)F(SEQ ID NO:19)和Och1(1212)XhoI R(SEQ ID NO:25)以及引物对KpnIOch1(+1)F(SEQ ID NO:26)和Och1(+801)BamHI R(SEQ ID NO:22)用于构建pUAH(+1)以便整合在OCH1编码区中紧邻终止密码子上游的位置(1212/+1)。
引物对SphIOch1(274)F(SEQ ID NO:19)和Och1(+3)XhoI R(SEQ ID NO:27)以及引物对KpnIOch1(+4)F(SEQ ID NO:28)和Och1(+801)BamHI R(SEQ ID NO:22)用于构建pUAH(+4)以便整合在OCH1 3’调控区中紧邻终止密码子下游的位置(+3/+4)。
引物对SphIOch1(274)F(SEQ ID NO:19)和Och1(+203)XhoI R(SEQ ID NO:29)以及引物对KpnIOch1(+204)F(SEQ ID NO:30)和Och1(+801)BamHI R(SEQ ID NO:22)用于构建pUAH(+204)以便整合在OCH1 3’调控区中的位置(+203/+204)。
引物对SphIOch1(-860)F(SEQ ID NO:31)和Och1(-110)XhoI R(SEQ ID NO:32)以及引物对KpnIOch1(-109)F(SEQ ID NO:33)和Och1(+641)BamHI R(SEQ ID NO:34)用于构建pUAH(-109)以便整合在OCH1 5’调控区起始密码子上游的位置(-110/-109)。
实施例3
OCH1基因中不同位置的打靶盒整合
图4A绘制了毕赤酵母的OCH1基因座中打靶整合位置。为进行整合,利用以下引物对,将一系列构建的OCH1打靶载体,包括pUAH(-109,1,554,1097,1166,+1,+4和+204)进行PCR扩增以产生线形形式的UAH(-109,1,554,1097,1166,+1,+4和+204)的OCH1打靶盒:
Och1(-709)F(SEQ ID NO:35)/Och1(491)R(SEQ ID NO:36)
Och1(-600)F(SEQ ID NO:37)/Och1(600)R(SEQ ID NO:38)
Och1(-47)F(SEQ ID NO:39)/Och1(1153)R(SEQ ID NO:40)
Och1(496)F(SEQ ID NO:41)/Och1(+484)R(SEQ ID NO:42)
Och1(565)F(SEQ ID NO:43)/Och1(+553)R(SEQ ID NO:44)
Och1(612)F(SEQ ID NO:45)/Och1(+600)R(SEQ ID NO:46)
Och1(615)F(SEQ ID NO:47)/Och1(+603)R(SEQ ID NO:48)
Och1(816)F(SEQ ID NO:49)/Och1(+803)R(SEQ ID NO:50)。
OCH1打靶盒含有URA3和ADE1表达盒,它们在DNA双链上,以相对取向彼此相邻定位。两种表达盒均侧接600bp相同长度的5’和3’整合同源序列(5’H和3’H),它们是基因特异性的同源序列以确保OCH1基因中打靶位置的精确整合。
利用MicroPulserTM电穿孔设备,按照生产商(BioRad,美国)的操作使用说明书,通过电穿孔将打靶盒转化入毕赤酵母营养缺陷型菌株JC301(ade1 his4 ura3)(Keck Graduate Institute)。转化的细胞在补充了20mg/L组氨酸的YNB平板上培养以选择腺嘌呤和尿嘧啶原养型。
图4B描述了打靶盒在毕赤酵母中紧邻OCH1基因的起始密码子上游的位置(-1/1)的代表性同源整合。在各转化板上,随机挑取菌落并培养以提取基因组DNA供PCR验证基因组的整合。两个引物对,P1(SEQ ID NO:51,位于基因组中5’同源区域的上游)/P2(SEQ ID NO:52,位于打靶片段的URA3内)和P3(SEQ ID NO:53,位于打靶片段的ADE1内)/P4(SEQ ID NO:54,位于基因组中3’同源区域的下游)用于验证打靶位置的同源整合(图4B)。图4C显示OCH1基因座中不同位置整合的PCR验证结果。通过P1/P2引物对分别扩增出预计的1300、2550和2550bp条带。这些菌株在利用P3/P4引物对的PCR中也扩增出预计的3500、2300和2300kb条带。PCR结果验证了相应的打靶片段分别成功整合在指定位置(-1/1,1212/+1,+3/+4)。亲代菌株JC301作为负对照,通过P1/P4引物对扩增1433bp条带,但通过P1/P2和P3/P4引物对没有扩增到条带。紧邻OCH1基因座的起始密码子上游整合的菌株称为och1(-1/+1,ADE1URA3)菌株。
本实施例证明,当利用600bp相同长度的同源序列时,OCH1基因中不同位置的同源重组整合效率显著不同(表1)。在编码区中这些位置(553/554,1096/1097,and 1165/1166)整合的整合转化株无法在100个筛选的菌落中鉴定到。由于在OCH1编码区的整合导致OCH1基因功能破坏并丧失细胞适应性,同源整合不是破坏基因组中OCH1编码区的优选机制。相反,打靶盒通过非同源末端连接随机整合入基因组。这与以前的报道一致,当利用约1kb或更长的同源序列时,毕赤酵母中OCH1的破坏效率显著较低,仅为0.1%,但其它试验室难以获得相同的结果(Choi,2003,Proc Natl Acad Sci U S A 100:5022–5027;Chen,2013,PLoS ONE 8(3):e57952)。
毕赤酵母在同源基因打靶中效率极低是众所周知的。同源重组基因替换事件的频率高度依赖于打靶片段的长度。当打靶同源序列低于500bp时,频率小于0.1%。
然而,与以前的报道不同的是,本发明发现,当利用600bp的短同源序列 时,紧邻终止密码子上游位置(1212/+1)的同源整合频率约为7%。这可能归结为OCH1功能的不完全破坏,因为整合导致OCH1基因转录生成OCH1无终止mRNA,其随后翻译生成具有一定功能活性的C-末端延伸OCH1产物。当形成的C-末端延伸产物保留一定活性时,邻近ORF的3’端位点的同源整合频率可能高于其它ORF区域中位点。
此外,本发明还发现在OCH1 5’-和3’-调控区中位置的同源重组的频率显著较高,例如在起始密码子上游的位置(-110/-109,-1/1)为40%、35%,终止密码子下游的两位置(+3/+4,+203/+204)为80%、25%。
实施例4
ADE1基因中不同位置的打靶盒整合
在ADE1基因中不同位置进行打靶盒整合以进一步验证OCH1基因打靶的结果。
图5A描述了基因组ADE1基因中打靶盒的整合位置。
图5B显示了通过PCR构建ADE1打靶盒的示意图。
PCR1,用毕赤酵母基因组DNA作为模板,用ADE1(-800)F(SEQ ID NO:55)和ADE1(-1)U R(SEQ ID NO:56)(该引物具有URA3重叠序列以供融合PCR)引物对作PCR扩增ADE1基因座的5’-同源序列(5’H,-800/-1);
PCR2,用pBLURA-SX载体作为模板,用A(-21)URA3F(SEQ ID NO:57)和URA3A(19)R(SEQ ID NO:58)(二者均具有ADE1重叠序列以供融合PCR)引物对扩增URA3表达盒。
PCR3,用毕赤酵母基因组DNA作为模板,用UADE1(1)F(SEQ ID NO:59)(该引物具有URA3重叠序列以供融合PCR)和ADE1(800)R(SEQ ID NO:60)引物对作PCR扩增ADE1基因的3'-同源序列(3’H,1/800)。
用ADE1(-800)F(SEQ ID NO:55)和ADE1(800)R(SEQ ID NO:60)引物对,通过重叠延伸PCR连接以上3种PCR产物(1,2,3)。如此产生了线形打靶盒UH(1),其整合在ADE1 5’调控区中紧邻起始密码子上游的位置(-1/1)。
以相同的方式,利用以下相应的引物对,通过PCR扩增和融合构建一系列ADE1打靶盒,它们整合在ADE1基因的不同位置:
引物对ADE1(-98)F(SEQ ID NO:61)和ADE1(703)U R(SEQ ID NO:62)(其具有URA3重叠序列以供融合PCR),引物对A(684)URA3F(SEQ ID NO:63)和URA3A(728)R(SEQ ID NO:64)(二者均具有ADE1重叠序列以供融合PCR)和引物对ADE1(704)F(SEQ ID NO:65)(其具有URA3重叠序列以供融合PCR)和ADE1(+591)R(SEQ ID NO:66)用于构建打靶盒UH(704)以便整合在ADE1编码区中的位置(703/704)。
引物对ADE1(62)F(SEQ ID NO:67)和ADE1(862)U R(SEQ ID NO:68)(其具有URA3重叠序列以供融合PCR)、引物对A(842)URA3F(SEQ ID NO:69)和URA3A(881)R(SEQ ID NO:70)(二者均具有ADE1重叠序列以供融合PCR)和引物对UADE1(863)F(SEQ ID NO:71)(其具有URA3重叠序列以供融合PCR)和ADE1(+750)R(SEQ ID NO:72)用于构建打靶盒UH(863)以便整合在ADE1编码区中的位置(862/863)。
引物对ADE1(62)F(SEQ ID NO:67)和ADE1(912)U R(SEQ ID NO:73)(其具有URA3重叠序列以供融合PCR)、引物对A(896)URA3 F(SEQ ID NO:74)和URA3A(+21)R(SEQ ID NO:75)(二者均具有ADE1重叠序列以供融合PCR)和引物对UADE1(+1)F(SEQ ID NO:76)(其具有URA3重叠序列以供融合PCR)和ADE1(+750)R(SEQ ID NO:72)用于构建打靶盒UH(+1)以便整合在编码区中紧邻ADE1终止密码子上游的位置(912/+1)。
引物对ADE1(62)F(SEQ ID NO:67)和ADE1(+3)U R(SEQ ID NO:77)(其具有URA3重叠序列以供融合PCR)、引物对A(896)URA3F(SEQ ID NO:78)和URA3A(+23)R(SEQ ID NO:79)(二者均具有ADE1重叠序列以供融合PCR)和引物对UADE1(+4)F(SEQ ID NO:80)(其具有URA3重叠序列以供融合PCR)和ADE1(+750)R(SEQ ID NO:72)用于构建打靶盒UH(+4)以便整合在3’调控区中紧邻ADE1终止密码子下游的位置(+3/+4)。
引物对ADE1(298)F(SEQ ID NO:81)和ADE1(+203)U R(SEQ ID NO:82)(其具有URA3重叠序列以供融合PCR)、引物对A(+186)URA3 F(SEQ ID NO:83)和URA3A(+226)R(SEQ ID NO:84)(二者均具有ADE1重叠序列以供融合PCR)和引物对ADE1(+204)F(SEQ ID NO:85)(其具有URA3重叠序列以供融合PCR)和ADE1(+1004)R(SEQ ID NO:86)用于构建打靶盒UH(+204)以便整合在ADE1 3’调控区中的位置(+203/+204)。
引物对ADE1(-895)F(SEQ ID NO:87)和ADE1(-110)U R(SEQ ID NO:88)(其具有URA3重叠序列以供融合PCR)、引物对A(-133)URA3 F(SEQ ID NO:89)和URA3A(-87)R(SEQ ID NO:90)(二者均具有ADE1重叠序列以供融合PCR)和引物对UADE1(-109)F(SEQ ID NO:91)(其具有URA3重叠序列以供融合PCR)和ADE1(691)R(SEQ ID NO:92)用于构建打靶盒UH(-109),以便整合在ADE1 5’调控区起始密码子上游的位置(-110/-109)。
这些线形打靶盒含有URA3表达基因,在两侧侧接长度相似在800bp左右(750-850)的5’和3’同源序列,这些序列是基因特异性同源序列以便精确整合在毕赤酵母ADE1基因的打靶位置。
通过电穿孔将这些打靶盒转化入毕赤酵母营养缺陷型菌株JC307(his4 ura3)(Keck Graduate Institute,USA)的细胞。转化的细胞生长在补加了20mg/L组氨酸的SC平板(8g/L SC不含组氨酸和尿嘧啶,20g/L葡萄糖,15g/L琼脂)上以选择尿嘧啶原养型。在2-3天培育期间,从平板随机挑取菌落以避免因白色/淡红色菌落而引起菌落挑取的偏差,因为ade1菌株中红色色素的累积以及淡红色菌落的出现需要较长时间的培育。从过夜培养菌落提取基因组DNA,用于通过PCR验证在ADE1基因中位置的整合。
引物对P5/P6(位于基因组中5’同源区上游,和3’同源区下游)用于验证基因组整合(图5B)。相应的P5/P6引物对进一步命名为P5-1(SEQ ID NO:93)/P6-1(SEQ ID NO:94)、P5-2(SEQ ID NO:95)/P6-2(SEQ ID NO:96)、P5-3(SEQ ID NO:97)/P6-3(SEQ ID NO:98)、和P5-4(SEQ ID NO:99)/P6-4(SEQ ID NO:100)以验证在ADE1基因中不同位置的整合。例如,成功扩增具有3763bp预期大小的条带表明在912/+1位置的染色体整合正确,但扩增大小为2398bp的条带表明无染色体整合(图5C)。
本实施例证明,当利用约800bp相似长度的同源序列时,ADE1基因中不同位点的同源重组整合效率显著不同(表1)。在邻近ORF 3’端的位置(862/863和912/+1)的整合频率超过15%。但在中部ORF位置(703/704)的整合频率仅有3%。与OCH1整合相似,邻近ORF 3’端位点的整合频率要高于其它ORF区域。
与OCH1整合结果一致,在ADE1 5’-和3’-调控区位置的同源整合频率显著 较高,例如在起始密码子上游的两位置(-110/-109,-1/1)为50%,65%,在终止密码子下游两位置(+3/+4,+203/+204)为30-45%。
以前的报道表明,同源重组基因打靶频率是基因依赖性的,例如OCH1基因具有极低的效率,而ADE1基因具有较高的频率。然而,本发明发现同源重组基因打靶频率主要取决于靶基因的区域。当利用小于1kb同源序列时,虽然同源重组整合在OCH1和ADE1的ORF区域以破坏基因功能具有完全不同的频率,但同源重组整合在OCH1和ADE1基因座的5’和3’区域都具有相似的高频率,频率超过25%。本发明发现在5’-和3’-调控区的同源重组基因整合的频率不依赖于基因,而且效率很高。此外,邻近ORF 3’端位点的基因整合频率高于其它ORF区域。基因组基因的不同区域中的同源重组打靶整合频率可以表示为以下次序:5’-调控区和3’-调控区﹥﹥编码区的3’端﹥其它编码区。本发明的这些发现为调控靶基因功能提供了新的机会。
实施例5
基因整合以调控β-半乳糖苷酶活性
虽然本发明已经确认在OCH1和ADE1的5’和3’调控区同源重组基因整合具有很高的效率,但是目前还没有系统分析来阐明5’和3’调控区的基因整合将会如何影响基因转录和蛋白质表达。
大肠杆菌(Escherichia coli)的lacZ基因编码β-半乳糖苷酶,其水解包括生色底物在内的各种β-D-半乳糖苷以产生有色产物。由于液体培养中β-半乳糖苷酶活性试验的便利性和灵敏度,其是常用的报道酶以监测基因表达的调控。通过融合lacZ ORF与基因的5’和3’调控区可以构建毕赤酵母的lacZ报道酶。
(1)构建5’AOX1-诱导的lacZ表达载体
图6和7描述了构建一系列5’AOX1-诱导的lacZ表达载体的示意图,其中URA3在lacZ ORF中毗邻起始和终止密码子以调控其表达。
PCR1,用大肠杆菌BL21(DE3)基因组DNA作为模板,用BamHIlacZ F(SEQ ID NO:101)(该引物具有BamH I限制性酶切位点)和lacZNotI R(SEQ ID NO:102)(该引物具有Not I和Xho I限制性酶切位点)引物对作PCR以扩增lacZ ORF(SEQ ID NO:128)。
PCR2,用大肠杆菌BL21(DE3)基因组DNA作为模板,利用BamHIlacZ F(SEQ ID NO:101)和lacZnsNotI R(SEQ ID NO:103)(该引物缺乏lacZ终止启动子并具有Not I和Xho I限制性酶切位点)引物对作PCR以扩增大肠杆菌lacZ ORF。
然后,分别用BamH I和Not I消化lacZ和lacZns的PCR产物。将酶切lacZ和lacZns的BamHI/NotI片段插入pPIC3.5K载体(Invitrogen)的BamH I和Not I位点以产生p5’AOX1-lacZ和p5’AOX1-lacZns载体(图6)。
PCR3,用pBLURA-SX载体作为模板,用BamHIURA3F(SEQ ID NO:104)(该引物具有BamH I限制性酶切位点)和URA3BamHI R(SEQ ID NO:105)(该引物具有BamH I限制性酶切位点)引物对作PCR以扩增毕赤酵母URA3表达盒。
用BamH I消化URA3表达盒的PCR产物,插入p5’AOX1-lacZ载体的BamH I位点。将含有两种取向的URA3的连接载体转化入大肠杆菌菌株Trans1-T1(TransGen Biotech,中国),进行菌落PCR以选择载体p5’AOX1-URA3-lacZ,其中URA3在同一链中,相同取向紧邻lac ORF上游(图6)。
PCR4,用pBLURA-SX载体作为模板,用NotIURA3 F(SEQ ID NO:106)(该引物具有Not I限制性酶切位点)和URA3NotI R(SEQ ID NO:107)(该引物具有Not I限制性酶切位点)引物对作PCR以扩增毕赤酵母URA3表达盒。
用Not I消化URA3表达盒的PCR产物,分别插入p5’AOX1-lacZ和p5’AOX1-lacZns载体的Not I位点。将含有两种取向的URA3的连接载体转化入噬菌体耐受性大肠杆菌菌株Trans1-T1,进行菌落PCR分别选出载体p5’AOX1-lacZ-URA3、p5’AOX1-lacZ-URA(-)、p5’AOX1-lacZns-URA3、p5’AOX1-lacZns-URA3(-)。p5’AOX1-lacZ-URA3和p5’AOX1-lacZns-URA3均含有URA3表达盒,其位于同一链中,相同取向紧邻lac ORF和lacZns ORF下游(图6)。另两个载体p5’AOX1-lacZ-URA3(-)和p5’AOX1-lacZns-URA3(-)含有URA3表达盒,其位于相对链中,相对取向紧邻lac ORF和lacZns ORF下游(图7)。
(2)构建5’OCH1-介导的lacZ表达载体
图8、9、10和11描述了构建一系列5’OCH1-介导的lacZ表达载体的示意图,其中URA3在lacZ ORF中毗邻起始和终止密码子以调控其表达。
PCR1,用基因组DNA作为模板,利用BamHIOCH1(-731)F(SEQ ID NO:108)(该引物具有BamH I限制性酶切位点)和OCH1(-1)L R(SEQ ID NO:109)(该引物 具有lacZ重叠序列以供融合PCR)引物对作PCR以扩增毕赤酵母OCH1的5’调控区(5’OCH1,-731/-1)。
PCR2,用大肠杆菌BL21(DE3)基因组DNA作为模板,用OLacZ F(SEQ ID NO:110)(该引物具有5’OCH1重叠序列以供融合PCR)和lacZXhoI R(SEQ ID NO:111)(该引物具有Xho I限制性酶切位点)引物对作PCR以扩增lacZ ORF。
PCR3,用大肠杆菌BL21(DE3)基因组DNA作为模板,用OLacZ F(SEQ ID NO:110)和lacZnsNotI R(SEQ ID NO:103)(该引物缺乏lacZ终止密码子并具有Not I和Xho I限制性酶切位点)引物对作PCR以扩增无终止密码子的lacZ ORF(lacZns)。
PCR4,用BamHIOCH1(-731)F(SEQ ID NO:108)和LacZXhoI R(SEQ ID NO:111)引物对,通过重叠-延伸PCR融合PCR1和PCR2产物。如此产生5’OCH1-lacZ的片段。
PCR5,用BamHIOCH1(-731)F(SEQ ID NO:108)和lacZnsNotI R(SEQ ID NO:103)引物对,通过重叠-延伸PCR融合PCR1和3产物。如此产生5’OCH1-lacZns的片段。
PCR6,用基因组DNA作为模板,用XhoIOCH1(+4)F(SEQ ID NO:112)(该引物具有Xho I限制性酶切位点)和OCH1(+798)SacI R(SEQ ID NO:113)(该引物具有Sac I限制性酶切位点)引物对作PCR以扩增毕赤酵母OCH1的3’调控区(3’OCH1,+4/+798)。
然后,用BamH I和Xho I消化5’OCH1-lacZ片段的PCR产物,用Xho I和Sac I消化3’OCH1的PCR产物。将5’OCH1-lacZ的BamHI-XhoI片段和3’OCH1的XhoI-SacI片段插入pBLHIS-SX载体的Sac I和BamH I位点以产生p5’OCH1-lacZ载体(图8)。
用BamH I和Xho I消化5’OCH1-lacZns片段的PCR产物,用Xho I和Sac I消化3’OCH1的PCR产物。将5’OCH1-lacZns的BamHI-XhoI片段和3’OCH1的XhoI-SacI片段插入pBLHIS-SX载体的Sac I和BamH I位点以产生p5’OCH1-lacZns载体(图8)。
PCR7,用基因组DNA作为模板,用BamHIOCH1(-731)F(SEQ ID NO:108)和OCH1(-1)U R(SEQ ID NO:114)(该引物具有URA3重叠序列以供融合PCR) 引物对作PCR以扩增OCH1的5’调控区(5’OCH1,-731/-1)。
PCR8,用pBLURA-SX载体作为模板,用OURA3 F(SEQ ID NO:115)(该引物具有OCH1重叠序列以供融合PCR)和URA3SphIXhoI R(SEQ ID NO:116)(该引物具有Sph I和Xho I限制性酶切位点)引物对作PCR以扩增毕赤酵母URA3表达盒。
PCR9,用BamHIOCH1(-731)F(SEQ ID NO:108)和URA3SphIXhoI R(SEQ ID NO:116)引物对,通过重叠-延伸PCR融合PCR7和8产物。如此产生5’OCH1-URA3的片段。
PCR10,用大肠杆菌BL21(DE3)基因组DNA作为模板,用SphILacZ F(SEQ ID NO:117)(该引物具有Sph I限制性酶位点)和LacZXhoI R(SEQ ID NO:111)引物对作PCR以扩增lacZ ORF。
然后,用BamH I和Xho I消化5’OCH1-URA3片段的PCR产物,用Sph I和Xho I消化lacZ ORF的PCR产物。将5’OCH1-URA3的BamHI-XhoI片段和lacZ的SphI-XhoI片段插入p5’OCH1-lacZ载体的BamHI和XhoI位点以产生p5’OCH1-URA3-lacZ载体(图9)。
PCR11,用pBLURA-SX载体作为模板,用XhoIURA3F(SEQ ID NO:3)(该引物具有Xho I限制性酶切位点)和URA3XhoI R(SEQ ID NO:10)(该引物具有Xho I限制性酶切位点)引物对作PCR以扩增毕赤酵母URA3表达盒。
用Xho I消化URA3表达盒的PCR产物,插入p5’OCH1-lacZ和p5’OCH1-lacZns载体的Xho I位点。将含有两种取向的URA3的插入载体转化入大肠杆菌菌株Trans1-T1,进行菌落PCR以分别选择载体p5’OCH1-lacZ-URA3、p5’OCH1-lacZ-URA3(-)、p5’OCH1-lacZns-URA3、p5’OCH1-lacZns-URA3(-)。p5’OCH1-lacZ-URA3和p5’OCH1-lacZns-URA3均含有URA3表达盒,其位于同一链中,相同取向紧邻lac ORF和lacZns ORF下游。另两个载体p5’OCH1-lacZ-URA3(-)和p5’OCH1-lacZns-URA3(-)均含有URA3表达盒,其位于相对链中,相对取向紧邻lacz ORF和lacZns ORF下游(图10和11)。
(3)lacZ表达载体的转化
用Sac I消化来线形化5’AOX1-诱导lacZ表达载体,包括p5’AOX1-lacZ、p5’AOX1-URA3-lacZ、p5’AOX1-lacZ-URA3、p5’AOX1-lacZ-URA3(-)、 p5’AOX1-lacZns-URA3和p5’AOX1-lacZns-URA3(-),并通过电穿孔将它们转化入毕赤酵母菌株GS115(his4)(Invitrogen)。将转化的细胞在YNB平板上培养以选择组氨酸原养型。根据生产商(Invitrogen)所述,将线形化的表达载体通过单交换(roll-in)重组整合在基因组中。
用stu I消化来线形化5’OCH1-介导lacZ表达载体,包括p5’OCH1-lacZ、p5’OCH1-URA3-lacZ、p5’OCH1-lacZ-URA3、p5’OCH11-lacZ-URA3(-)、p5’OCH1-lacZns-URA3和p5’OCH1-lacZns-URA3(-),并通过电穿孔将它们转化入毕赤酵母菌株GS115。将转化的细胞在YNB平板上培养以选择组氨酸原养型。将线形化的表达载体通过单交换(roll-in)重组整合在his4基因处。
(4)lacZ mRNA的实时PCR分析
将5’AOX1-诱导lacZ表达载体的转化细胞在5ml BMGY培养基(10g/L酵母提取物、20g/L蛋白胨、13.4g/L YNB不含氨基酸、100mM磷酸钾缓冲液、pH6.0、0.4mg/L生物素、10ml/L甘油)中,30℃,以225rpm振荡培养48小时。以3000g离心5分钟沉淀细胞,然后将沉淀的细胞重悬在5ml BMMY培养基(10g/L酵母提取物、20g/L蛋白胨、13.4g/L YNB不含氨基酸、100mM磷酸钾缓冲液、pH 6.0、0.4mg/L生物素、10ml/L甲醇),30℃,225rpm振荡以诱导lacZ表达。用50μl 100%甲醇(1%终浓度)每日两次掺入培养液,将诱导再维持48小时。随后,以3000g离心细胞10分钟。用5ml水洗涤,再离心以收集细胞沉淀。将细胞沉淀用于β-半乳糖苷酶试验,或保存在-80℃用于总RNA分离。
将5’OCH1-介导lacZ表达载体的转化细胞在5ml YPD培养基中,30℃,以225rpm振荡培养72小时。随后,以3000g离心细胞10分钟,用5ml水洗涤,再离心以收集细胞沉淀。将细胞沉淀用于β-半乳糖苷酶试验,或保存在-80℃用于总RNA分离。
按照生产商的使用说明,利用试剂(Lifetechnologies)进行总RNA分离。
按照生产商的使用说明,利用ReverTra Ace-α-第一链cDNA合成试剂盒(Toyobo)进行RNA的逆转录。
实时PCR反应:10μl 2×iTaqTM UniversalGreen supermix(BioRad,Hercules,CA),1μl cDNA和100nM各自的GAPDH F(SEQ ID NO:118)/R(SEQ ID NO:119)和LacZ F(SEQ ID NO:120)/R(SEQ ID NO:121)引物,20μl总反应体积。利用LightCycler LC480(Roche),以如下参数进行PCR反应:1轮95℃、1分钟,40轮95℃、10秒,58℃、10秒,72℃、10秒。所有的样品一式三份,并测试数次。利用生产商(Roche)的软件分析实时PCR数据。采用比较CT方法(ΔΔCT方法)测定mRNA的相对表达。甘油醛-3-磷酸脱氢酶用作内源性对照以便定量测定基因表达。
图12A和B显示当URA3表达盒整合于起始和终止密码子附近时对5’AOX1和5’OCH1启动的lacZ mRNA表达的抑制作用。当URA3表达盒在同一链中,以相同取向紧邻lacZ ORF起始密码子的上游整合时,其将lacZ mRNA水平分别有效降低60%和70%。这种mRNA降低可归结为3’URA3终止子,其位于lacZ ORF上游并阻断5’AOX1或5’URA3启动子启动的lacZ转录。但是3’URA3终止子未能完全终止转录而产生低水平的异常lacZ mRNA,其缺乏合适的5’UTR以供翻译。
当URA3表达盒在DNA双链中,以两种取向紧邻lacZ ORF终止密码子的上游整合时,预计能产生缺乏终止密码子的异常lacZ mRNA(无终止lacZ mRNA,lacZns)。当URA3表达盒在DNA双链中,以两种取向紧邻lacZ ORF终止密码子的下游整合时,预计能产生含有异常3’UTR的lacZ mRNA。RT-PCR分析显示终止密码子附近的整合能增加或降低5’AOX1或5’OCH1启动的lacZ异常mRNA水平(图12A和B)。这些结果与以前的报道不一致,即,细胞具备监督系统来识别和消除异常mRNA以避免产生可能有害的蛋白质产物(van Hoof A,Frischmeyer PA,Dietz HC,Parker R(2002)Exosome mediated recognition and degradation of mRNAs lacking a termination codon.Science 295:2262–2264)。因此,必需要评估DNA双链中、以两种取向在终止密码子附近的整合以获得调节靶基因转录的最佳效应。
(5)β-半乳糖苷酶活性试验
为进一步了解对5’AOX1和5’OCH1介导的蛋白表达的抑制作用,采用标准方案检测收集的细胞沉淀中β-半乳糖苷酶的胞内比活性(Ausubel F M et al.Current Protocols in Molecular Biology,Wiley InterScience,2010)。
图13A和B显示对应于URA3表达盒整合在起始和终止密码子附近时,β-半乳糖苷酶的相对胞内比活性。当URA3在同一链中,以相同取向整合在紧邻lacZ ORF起始密码子上游的位置时,细胞中没有可检测的β-半乳糖苷酶活性。β-半乳糖苷酶活性的完全抑制可归结于转录和翻译调控作用。首先,3’URA3的终止作用显著降低异常缺乏5’UTR的lacZ mRNA转录。其次,在不含合适5’UTR的异常lacZ mRNA中无法启动翻译。
5’调控区,特别是紧邻ORF上游位置的基因整合能通过阻遏转录和翻译来专一性抑止靶基因表达。目前调控基因转录的方法主要包括:通过抑止分子与DNA结合改变模扳功能,通过抑止分子与RNA聚合酶结合而抑止其转录活性。调控蛋白翻译的方法主要采用小RNA和RNA-结合蛋白与mRNA结合来改变其翻译能力。相比之下,基因组基因中,特别是ORF上游的基因整合是控制靶基因表达的更为有效的方法。
由于URA3整合对lacZ异常mRNA水平有不同作用,在不同链中、以不同取向在终止密码子附近的URA3整合对细胞中5’AOX1和5’OCH1介导的β-半乳糖苷酶翻译有各种不同抑制作用。当URA3以合适取向在终止密码子附近整合时,β-半乳糖苷酶活性可降低最多70%。这种抑制作用比以前报道的通过microRNA抑制更有效。据报道,microRNA对mRNA的稳定性和翻译水平只能抑制32%和4%(Noah Spies,Christopher B.Burge,and David P.Bartel(2013).3’UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts,Genome Research,23:2078–2090)。
本发明显示了通过在终止密码子附近整合来控制靶基因表达的另一种方法。必需评估DNA双链中、以两种取向在终止密码子附近的基因整合以获得最佳调节结果。
实施例6
基因整合以抑制OCH1表达
为进一步了解基因整合对调节基因组中基因转录的作用,选择实施例3中的代表性整合菌株来分析OCH1基因的mRNA转录。将对照菌株JC307和3种整合菌株och1(-1/+1)、(1212/+1)、(+3/+4)的细胞在5ml YPD培养基中,30℃,以225rpm振荡培养72小时。随后,以3000g离心细胞10分钟,用5ml水洗涤,再离心以收集细胞沉淀,并保存在-80℃用于总RNA分离。
利用试剂(Lifetechnologies)进行总RNA分离,利用ReverTra Ace-α-第一链cDNA合成试剂盒(Toyobo)进行cDNA的逆转录。
实时PCR反应:10μL 2×iTaqTM UniversalGreen supermix(BioRad,Hercules,CA),1μl cDNA和100nM各自的GAPDH F(SEQ ID NO:118)/R(SEQ ID NO:119)和OCH1F(SEQ ID NO:122)/R(SEQ ID NO:123)引物,20μL总反应体积。利用LightCycler LC480(Roche),以如下参数进行PCR反应:1轮95℃、1分钟,40轮95℃、10秒,58℃、10秒,72℃、10秒。所有的样品一式三份,并测试数次。利用生产商(Roche)的软件分析实时PCR数据。采用比较CT方法(ΔΔCT方法)测定mRNA的相对表达。甘油醛-3-磷酸脱氢酶(GAPDH)用作内源性对照以便定量测定基因表达。
图14显示了这些菌株中的OCH1 mRNA相对表达量。起始密码子上游位置的基因整合能有效抑制OCH1 mRNA水平超过90%。终止密码子下游位置的基因整合也能降低OCH1 mRNA水平。然而,终止密码子上游位置的基因整合显著增加OCH1 mRNA水平。该结果类似于在实施例5中lacZ mRNA调节的观察结果。
实施例7
OCH1抑制菌株中的蛋白质糖基化
N-糖基化是将寡糖连接于共有序列Asn-X-Ser/Thr内的特定天冬酰胺残基(Kornfeld,R.& Kornfeld,S.(Assembly of asparagine-linked oligosaccharides.Annu.Rev.Biochem.54,631–664,1985)。简言之,在真核细胞中,Glc3Man9GlcNAc2寡糖在内质网(ER)膜的腔侧转移到初生多肽链。利用葡糖苷酶I和II除去3个葡萄糖残基后,通过甘露糖苷酶I除去一特定末端的α-1,2-甘露糖。这些反应在大多数低级和高级真核生物之间是保守性的。此时,正确折叠的Man8GlcNAc2 N-糖基化蛋白离开ER到达高尔基体复合物,在该处糖链经历进一步的物种和细胞类型特异性加工。
高级真核生物中,ER的Man8GlcNAc2糖链被几种α-1,2-甘露糖苷酶进一步处理以产生Man5GlcNAc2 N-聚糖,相比之下,酵母菌通过将α-1,6-甘露糖残基加入三甘露糖基核心的α-1,3-甘露糖来修饰Man8GlcNAc2糖链,该反应由高尔 基体中的Och1p催化。然后几种(磷酸)甘露糖基转移酶进一步延长这种“启动甘露糖”。如此导致高甘露糖链结构Man9-100GlcNAc2,其由最多数十α-1,6-甘露糖的骨架与短的侧链构成。这种不同与人蛋白糖链的N-高甘露糖链极大地降低蛋白质在体内半衰期,其具有免疫原性并有碍下游工艺操作。
为阻断N-高甘露糖链的形成,目前已进行了大量的工作以敲除酵母菌基因组中的OCH1基因。然而,同源基因打靶直接破坏OCH1基因编码区及其功能的效率极低。在本发明中,5’OCH1调控区的基因整合可应用于阻断重组蛋白产物中Och1p启动的高甘露糖基化过程。
由Generay合成密码子优化的小鼠白介素-22(mIL-22,针对酵母菌密码子作优化的含his-标签小鼠IL-22成熟肽的DNA序列如SEQ ID NO:129所示)做模板,用MIL22F(SEQ ID NO:124)/R(SEQ ID NO:125)引物对作PCR扩增。用Xho I和Not I限制性酶消化PCR产物,并克隆入pPICZα(Invitrogen)的Xho I/Not I位点以产生mIL-22表达载体,其能表达并分泌含His-标签的mIL-22。用限制性酶Sac I线形化该表达载体,并电穿孔转入GS 115和och1(-1/+1)菌株。转化的细胞在补加了100mg/L的Zeocin的YPD平板上培养。根据生产商(Invitrogen)所述,通过单交换(roll-in)重组将线形化的载体整合在AOX1基因座。
将转化的细胞在5ml YPD培养基中,30℃,225rpm振荡培养24小时。以3000g离心沉淀细胞5分钟,重悬在5ml BMGY培养基中,30℃,225rpm振荡培养24小时。然后,以3000g离心沉淀细胞5分钟,重悬在5ml BMGY培养基中,30℃,225rpm振荡培养以诱导mIL-22表达。用50μl 100%甲醇(1%终浓度)每日两次掺入培养液,将诱导再维持72小时。随后,通过离心收获培养上清液(3000g,10分钟),-20℃冷冻上清液待用。
按照生产商的使用说明书(南京金斯瑞生物科技有限公司),通过Ni-亲和层析从上清液中纯化带His-标签的mIL-22蛋白。
采用以前报道的方法(Gregg JM(2010)Pichia Protocols,Second edition.Totowa,New Jersey:Humanna Press),通过N-糖苷酶F(PNGaseF)(New England Biolabs,Beverly,MA)处理从带His-标签的mIL-22蛋白释放并分离聚糖。
按照生产商的使用说明书,利用Ultraflex MALDI-TOF(bruker daltonics,不来梅,德国)质谱仪测定聚糖的分子量。
图15A显示GS115菌株中从mIL-22释放的N-糖链的质谱图。其显示主要的N-高甘露糖链为Man14GlcNAc2(m/z:2695.0,[M+H]+)。图15B显示och1(-1/+1)菌株中从mIL-22释放的N-糖链的质谱图。其显示没有Man14GlcNAc2的N-高甘糖链。主要的N-糖链出现在m/z为1843.6处,在Man8GlcNAc2(m/z:1720,[M+H]+)和Man9GlcNAc2(m/z:1883,[M+H]+)之间。这可能通过修饰Man8GlcNAc2来产生这种主要的N-糖链,其需要进一步的确认。N-糖链分析表明OCH1ORF上游的基因整合能有效阻断Och1p启动的高甘露糖基化过程。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!