一种醋酸地加瑞克制备后处理方法与流程
本发明涉及一种醋酸地加瑞克的制备后处理方法。
背景技术:
美国FDA批准Ferring Pharmaceuticals公司degarelix粉针(Firmagon)上市,用于治疗晚期前列腺癌。化学名: N-乙酰基-3-(2-萘基)-D-丙氨酰 -4-氯-D-苯丙氨酰-3-(3-吡啶基)-D-丙氨酰-L丝氨酰-4-[[[(4S)-六氢-2,6-二氧代-4-嘧啶基]羰基]氨基]-L-苯丙氨酰-4-[(氨基羰基)氨基]-D苯丙氨酰-L-亮氨酰-N6-(1-甲基乙基)-L赖氨酰-L-脯氨酰- D-丙氨酰胺,结构如下:
Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-Hor)-D-4Aph(Cbm)-Leu-Lys(iPr)-Pro-D-Ala-NH2。
其中Ac是乙酰基,2Nal是2-萘基丙氨酸,4Cpa是4-氯苯丙氨酸,3Pal是3-吡啶基丙氨酸,Ser是丝氨酸,4Aph是4-氨基苯丙氨酸,Hor是氢化乳清酸,Cbm是氨基甲酰胺,Leu是亮氨酸,Lys(iPr)是N6-异丙基赖氨酸,Pro是脯氨酸而Ala是丙氨酸。
本品系一促性腺激素释放激素(GnRH)受体抑制剂类药物,可逆性抑制垂体GnRH受体来减少促性腺激素释放继而抑制睾酮的释放。本品通过抑制对前列腺癌持续生长至关重要的睾酮来延缓前列腺癌的生长和恶化。以激素治疗前列腺癌来降低睾酮浓度的初期会造成睾酮浓度激增,此初始刺激该激素受体可暂时性促进肿瘤生长而不是抑制它,而地加瑞克则不会。Ⅲ期临床研究显示,本品降低睾酮浓度的效果至少可与亮丙瑞林储库型控释注射剂(Lupron Depot)相媲美,而且降低睾酮浓度在统计学上显著快。在治疗的第3日,本品组96%达到去生殖腺的睾酮浓度,亮丙瑞林组效果为0%。第14日,本品组99%达到去生殖腺的睾酮浓度,亮丙瑞林组为18%。在临床研究中,前列腺特异抗原(PSA)浓度可作为监测的第2个疗效判断终点。使用degarelix 2周后降低PSA 64%,1月后85%,3月后95%,在治疗的整个1年中始终抑制PSA。
本品剂量规格:degarelix 80 mg、120 mg/瓶。该剂量规格在多肽品种较高,所以急需一种中间体易储存,产品更稳定,易于放大,适合规模化生产的制备方法。
中国专利CN102428097介绍了一种醋酸地加瑞克原料药的制备方法和CN102204889介绍了一种醋酸地加瑞克冻干粉针剂的制备方法,均未涉及到地加瑞克在原料药及后续制剂生产过程中极易产生的自聚集成凝胶现象,因此并未对地加瑞克的原料及制剂的粘度进行相应的检测与控制。
中国专利CN104334182公开了一种地加瑞克的制作方法,具体步骤为:纯化通过液相或固相肽合成所获得的地加瑞克以获得具有至少95%纯度的地加瑞克溶液;蒸发溶剂以浓缩所述地加瑞克溶液以获得聚集的地加瑞克;将聚集的地加瑞克用乙酸解聚;冻干解聚的地加瑞克以提供所述地加瑞克药物物质。
现有的地加瑞克制备过程中,由于地加瑞克的物理化学特性,极易在水溶液中自聚集,并且最终形成凝胶水溶液。30%醋酸解聚样品需要更低温(<-20℃)才能冻实样品,冻干制冷温度要求低且冻干效果不是很好。因此对于批量化工业生产中,地加瑞克中间体的存放及产品质量的稳定性都存在较大影响。本发明人研究发现,虽然有些方法可以解聚自聚集的地加瑞克,但加入大量醋酸的同时会增加冻干难度并影响冻干效果,而长时间地加瑞克水溶液浓缩过程中也容易造成地加瑞克的稳定性变差,从而使终产品质量水平及收率降低。为此,本发明人重点研究如何解决地加瑞克自聚集问题的同时而不影响地加瑞克产品的质量及收率问题。
本发明人用现有的制备方法均无法有效防止地加瑞克纯化后水溶液自聚集现象产生,即使能够解聚也对冻干设备要求高且难以保证冻干效果。为此,本发明人对地加瑞克的制备方法进行了研究,从而得到了本发明的技术方案,使得醋酸地加瑞克在生产过程中不产生自聚集,产品稳定,易于冻干,最终产品纯度高,适合规模化生产。
技术实现要素:
本发明的目的是提供一种高效率、高纯度以及低杂质的地加瑞克的制备后处理方法。本发明需要解决的技术问题是:选择一种地加瑞克的制备方法,解决(1)制备方法过程中自聚集现象,(2) 产品纯度不高,(3) 冻干制冷温度要求高,(4)工艺不稳定。
本发明的技术方案为:
使用在地加瑞克中间体每次旋蒸前添加少量醋酸,并采用分批旋蒸冷冻后复融,再混样后合并冻干的工艺,可在规模化生产中有效解决地加瑞克自聚集问题。
为此本发明提供一种地加瑞克的制备后处理方法,其特征在于,步骤如下:
步骤1,通过对固相合成法所获得的地加瑞克进行纯化,从而获得纯度大于99.0%的地加瑞克溶液 ;
步骤2,取转盐后待旋蒸处理的地加瑞克溶液,加入5~20%的醋酸,后搅拌均匀;
步骤3,将步骤2所得溶液分批次在30~35℃水浴条件下进行旋蒸浓缩除去有机改性剂,使得地加瑞克溶液的浓度达到30~50g/l;
步骤4, 将步骤3所得浓缩液均置于-20℃下冷冻保存,待所有批次步骤2溶液旋蒸浓缩完后将所有冷冻保存的旋蒸浓缩液均置于20~30℃水浴下进行复融后合并;
步骤5,将步骤4所得合并溶液经过冷冻干燥得到纯品醋酸地加瑞克。
优选的操作步骤如下:
步骤1,纯化通过固相合成法所获得的地加瑞克,从而获得纯度大于99.0%的地加瑞克溶液 ;
步骤2,取转盐后待旋蒸处理的地加瑞克溶液,加入5%的醋酸,后搅拌均匀;
步骤3,将步骤2所得溶液分批次在30~35℃水浴条件下进行旋蒸浓缩除去有机改性剂,使得地加瑞克溶液的浓度达到50g/l;
步骤4, 将步骤3所得浓缩液均置于-20℃下冷冻保存,待所有批次步骤2溶液旋蒸浓缩完后将所有冷冻保存的旋蒸浓缩液均置于20℃水浴下进行复融后合并;
步骤5,将步骤4所得合并溶液经过冷冻干燥得到醋酸地加瑞克原料药。
以下通过实验数据进一步说明本发明的有益效果:
采用中国专利CN104334182方法制备的醋酸地加瑞克,精肽纯度达到了97.50%,收率为29%,醋酸地加瑞克的粘度为2.3mPas;
采用本发明的方法制备的醋酸地加瑞克,精肽纯度达到了99.76%,收率为53.2%,醋酸地加瑞克的粘度为2.5mPas。
本发明的方法是经过筛选获得的,筛选过程如下:
优选实验一:
加入醋酸的比例选择:不加入;5%;10%;20%
为此提出了4种实验条件:
实验条件1:转盐后溶液不添加醋酸;
实验条件2:转盐后溶液添加5%醋酸;
实验条件3:转盐后溶液添加10%醋酸;
实验条件4:转盐后溶液添加20%醋酸。
采用实验条件1的方法制备地加瑞克,在旋蒸的过程中,醋酸地加瑞克溶液开始自聚集,并最终形成凝胶,无法冻干;
采用实验条件2的方法制备地加瑞克,旋蒸浓缩完后,冻干得到精肽纯度达到了99.70%,收率为52%,醋酸地加瑞克的粘度为2.4mPas;
采用实验条件3的方法制备地加瑞克,旋蒸浓缩完后,冻干得到精肽纯度达到了99.70%,收率为50%,醋酸地加瑞克的粘度为2.4mPas;
采用实验条件4的方法制备地加瑞克,旋蒸浓缩完后,冻干中醋酸地加瑞克溶液不易结晶冻实,较难冻干,对于冻干设备存在较高要求。
根据以上实验现象,优选旋蒸浓缩前加入醋酸的量为转盐后醋酸地加瑞克溶液体积的5%~10%。
优选实验二:
转盐溶液旋蒸温度,时间及醋酸地加瑞克冻干前浓度选择:
温度:30℃;35℃
时间:2h;4h
浓度:30g/l;50g/l
为此提出了4种实验条件:
实验条件5:转盐后溶液添加5%醋酸,在30℃下旋蒸2小时,使醋酸地加瑞克溶液的浓度为30g/L;
实验条件6:转盐后溶液添加5%醋酸,在30℃下旋蒸4小时,使醋酸地加瑞克溶液的浓度为50g/L;
实验条件7:转盐后溶液添加5%醋酸,在35℃下旋蒸2小时,使醋酸地加瑞克溶液的浓度为30g/L;
实验条件8:转盐后溶液添加5%醋酸,在35℃下旋蒸4小时,使醋酸地加瑞克溶液的浓度为50g/L。
采用实验条件5的方法制备地加瑞克,旋蒸浓缩完后,冻干得到精肽纯度达到了99.70%,收率为52%,醋酸地加瑞克的粘度为2.4mPas;
采用实验条件6的方法制备地加瑞克,旋蒸浓缩完后,冻干得到精肽纯度达到了99.68%,收率为51%,醋酸地加瑞克的粘度为2.7mPas;
采用实验条件7的方法制备地加瑞克,旋蒸浓缩完后,冻干得到精肽纯度达到了99.71%,收率为52%,醋酸地加瑞克的粘度为2.1mPas;
采用实验条件8的方法制备地加瑞克,旋蒸浓缩完后,冻干得到精肽纯度达到了99.65%,收率为51%,醋酸地加瑞克的粘度为2.4mPas。
根据以上实验现象,结合实际工艺需求,优选在温度为30~35℃,旋蒸2~4h,使得醋酸地加瑞克的浓度为50g/L。
优选实验三:
复融温度,时间选择:
温度:20℃;30℃
时间:2h;4h
为此提出了2种实验条件:
实验条件9:将分批预冻的浓缩后溶液均置于20℃的水浴中,复融4h后,合并溶液,进行冻干;
实验条件10:将分批预冻的浓缩后溶液均置于30℃的水浴中,复融2h后,合并溶液,进行冻干;
采用实验条件9的方法制备地加瑞克,冻干得到精肽纯度达到了99.67%,收率为51%,醋酸地加瑞克的粘度为2.3mPas;
采用实验条件10的方法制备地加瑞克,冻干得到精肽纯度达到了99.66%,收率为51%,醋酸地加瑞克的粘度为2.3mPas。
根据以上实验现象,优选复融温度为20~30℃,复融时间为2~4h。
本发明的有益效果是:提供了一种高纯度(99.76%)、高收率(50%以上)以及成品粘度可控的醋酸地加瑞克制备后处理方法。
附图说明
图1 专利CN104334182方法制备后醋酸地加瑞克HPLC检测图谱;
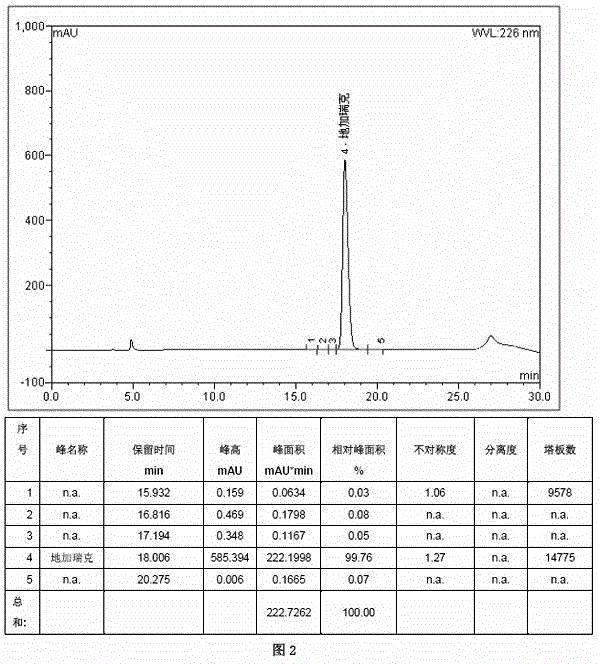
图2 本发明方法制备后醋酸地加瑞克HPLC检测图谱(1mmol);
图3 本发明方法制备后醋酸地加瑞克HPLC检测图谱(300mmol)。
具体实施方式
实施例1:
(1)取2.0g(1.0mmol)的Rink amide MBHA (取代度=0.50mmol/g)树脂加入到反应柱中,加入15mlDCM溶胀30min,去保护:加入15ml的V哌啶:VDMF =20:80混合溶液,搅拌反应5min后抽去,再次加入上述混合溶液,搅拌反应10min后抽去,如此进行2次脱保护,然后用DMF洗涤6次,每次15mL。
(2)偶联Fmoc-D-Ala-OH:将0.94g (3.0mmol) Fmoc-D-Ala-OH和0.51g (3.0mmol) 6-Cl-HOBt用溶剂DMF溶解,冰浴冷却温度为0~10℃,加入465μl (3.0mmol) DIC,激活5分钟后加入到反应柱中与脱完保护的载体进行反应,25~35℃下反应2小时,抽除液体,DMF洗涤3次,每次15mL。
(3)重复偶联:重复去除Fmoc保护基及接肽反应步骤的操作,按照地加瑞克氨基酸序列,依次偶联:Fmoc-Pro-OH、Fmoc-Ilys(Boc)-OH、Fmoc-Leu-OH、Fmoc-D-4Aph(Dde)-OH、Fmoc-4Aph(Teoc)-OH、Fmoc-Ser(tBu)-OH、Fmoc-D-3Pal-OH、Fmoc-D-Phe(4Cl)-OH、Fmoc-D-2Nal-OH。
(4)醋酐封端:先去保护基Fmoc,加入15ml的V哌啶:VDMF =20:80混合溶液,搅拌反应5min后抽去,再次加入上述混合溶液,搅拌反应10min后抽去,如此进行2次脱保护,然后用DMF洗涤6次,每次15mL。取1ml醋酐、1ml吡啶和1mlDMF,冰浴冷却,缓慢加入载体中,25~35℃下反应2小时,抽除液体,用DMF洗涤6次,每次15mL。
(5)去Dde保护基:加入15ml的V水合肼:VDMF =2:98混合溶液,搅拌反应5min后抽去,再次加入上述混合溶液,搅拌反应10min后抽去,如此进行2次脱保护,然后用DMF洗涤6次,每次15mL。
(6)偶联Cbm:加入270μl(2mmol)三甲基硅基异氰酸酯的DMF溶液,冰浴冷却,缓慢加入载体中,低温反应2小时,25~35℃下反应24小时,抽除液体,用DMF洗涤3次,每次15mL。
(7)去Teoc保护基:加入1.04g(4mmol)四丁基氟化铵的DMF混合溶液,25~35℃下反应2小时,抽除液体,用DMF洗涤3次,每次15mL。
(8)偶联L-Hor:将0.73g (3.0mmol) L-Hor-OH和0.51g (3.0mmol) 6-Cl-HOBt用溶剂DMF溶解,冰浴冷却温度为0~10℃,加入465μl (3.0mmol) DIC,激活5分钟后加入到反应柱中与脱完保护的载体进行反应,25-35℃下反应2小时,抽除液体,DMF洗涤3次,每次15mL。
(9)裂解:将上述肽树脂用15ml VTIS:VPhSMe:VTFA=5:5:90裂解液裂解,裂解时间:2小时,过滤,旋蒸,旋蒸母液加入90ml的乙醚中,沉降后离心洗涤5次,每次45mL。制得地加瑞克粗肽1.72g。
(10)第一次纯化:将色谱柱用50%乙腈冲洗干净后用5%乙腈平衡系统5min,以1-3g/次的上样量进行HPLC纯化,流动相:A为0.1%TFA水溶液;流动相B为乙腈,梯度程序为:初始状态流动相B为10%,保持5分钟,然后在60分钟内将流动相B比例增至50%,流速为:20 mL/min。于226nm波长处检测,根据色谱峰分段收集馏分,并将收集的馏分用分析液相色谱仪进行检测,纯度≥99.0%且单杂≤0.2%馏分即为第一步合格馏分。其余为不合格馏分,不合格馏分中纯度大于80%部分再进行回收纯化,纯度小于80%舍弃。
(11)第二次纯化:将色谱柱用50%乙腈冲洗干净后用5%乙腈平衡系统5min,以1-3g/次的上样量进行HPLC纯化,流动相:A为0.3%醋酸水溶液,流动相B为乙腈,梯度程序为:初始状态流动相B为10%,保持5分钟,然后在60分钟内将流动相B比例增至50%,流速为:20 mL/min。于226nm波长处检测,根据色谱峰分段收集馏分,并将收集的馏分用分析液相色谱仪进行检测,纯度≥99.5%且单杂≤0.1%馏分即为第二步合格馏分。其余为不合格馏分,不合格馏分中纯度大于80%部分再进行回收纯化,纯度小于80%舍弃。
(12)转盐的条件:流 动 相:A相:50 mmol/L乙酸铵的水溶液:乙腈=95:5;B相:0.1 %醋酸的水溶液:乙腈=95:5;C相:0.1 %醋酸的水溶液:乙腈=50:50,梯度程序为:以流动相A等梯度洗脱20分钟, 转换成流动相B等梯度洗脱10分钟, 转换成流动相C等梯度洗脱25分钟。以1-3g/次的上样量进行转盐,按照上述梯度要求进行洗脱,于226nm波长处检测,将有紫外吸收值的样品收集合并。
(13)浓缩及冻干:转盐后合并馏分加入5%的乙酸,分两批分别旋蒸浓缩1h,至地加瑞克浓度为50mg/ml左右,每批旋蒸完后均放入冰箱于-20℃下冷冻保存,待冻干前,将两批浓缩液均置于20℃水浴复融2h后合并,冻干后得到醋酸地加瑞克精肽:0.90g,纯度:99.76%,收率:53.2%。醋酸地加瑞克的粘度为2.5mPas。
实施例2:
(1)取600.0g(300.0mmol)的Rink amide MBHA (取代度=0.50mmol/g)树脂加入到反应柱中,加入4000mlDCM溶胀30min,去保护:加入4000ml的V哌啶:VDMF =20:80混合溶液,搅拌反应5min后抽去,再次加入上述混合溶液,搅拌反应10min后抽去,如此进行2次脱保护,然后用DMF洗涤6次,每次4000mL。
(2)偶联Fmoc-D-Ala-OH:将283.6g (900.0mmol) Fmoc-D-Ala-OH和153.6g (900.0mmol) 6-Cl-HOBt用溶剂DMF溶解,冰浴冷却温度为0~10℃,加入144ml (900.0mmol) DIC,激活5分钟后加入到反应柱中与脱完保护的载体进行反应,25-35℃下反应2小时,抽除液体,DMF洗涤3次,每次4000mL。
(3)重复偶联:重复去除Fmoc保护基及接肽反应步骤的操作,按照地加瑞克氨基酸序列,依次偶联:Fmoc-Pro-OH、Fmoc-Ilys(Boc)-OH、Fmoc-Leu-OH、Fmoc-D-4Aph(Dde)-OH、Fmoc-4Aph(Teoc)-OH、Fmoc-Ser(tBu)-OH、Fmoc-D-3Pal-OH、Fmoc-D-Phe(4Cl)-OH、Fmoc-D-2Nal-OH。
(4)醋酐封端:先去保护基Fmoc,加入4000ml的V哌啶:VDMF =20:80混合溶液,搅拌反应5min后抽去,再次加入上述混合溶液,搅拌反应10min后抽去,如此进行2次脱保护,然后用DMF洗涤6次,每次4000mL。取300ml醋酐、300ml吡啶和300mlDMF,冰浴冷却,缓慢加入载体中,25-35℃下反应2小时,抽除液体,用DMF洗涤6次,每次4000mL。
(5)去Dde保护基:加入4000ml的V水合肼:VDMF =2:98混合溶液,搅拌反应5min后抽去,再次加入上述混合溶液,搅拌反应10min后抽去,如此进行2次脱保护,然后用DMF洗涤6次,每次4000mL。
(6)偶联Cbm:加入84ml(600mmol)三甲基硅基异氰酸酯的DMF溶液,冰浴冷却,缓慢加入载体中,低温反应2小时,25~35℃下反应24小时,抽除液体,用DMF洗涤3次,每次4000mL。
(7)去Teoc保护基:加入312g(1200mmol)四丁基氟化铵的DMF混合溶液,25~35℃下反应2小时,抽除液体,用DMF洗涤3次,每次4000mL。
(8)偶联L-Hor:将220.2g (900.0mmol) L-Hor-OH和153.6g (900.0mmol) 6-Cl-HOBt用溶剂DMF溶解,冰浴冷却温度为0~10℃,加入144ml (900.0mmol) DIC,激活5分钟后加入到反应柱中与脱完保护的载体进行反应,25~35℃下反应2小时,抽除液体,DMF洗涤3次,每次4000mL。
(9)裂解:将上述肽树脂用4500ml VTIS:VPhSMe:VTFA=5:5:90裂解液裂解,裂解时间:2小时,过滤,旋蒸,旋蒸母液加入9000ml的乙醚中,沉降后离心洗涤5次,每次13500mL。制得地加瑞克粗肽534g。
(10)第一次纯化:将色谱柱用50%乙腈冲洗干净后用5%乙腈平衡系统5min,以20-35g/次的上样量进行HPLC纯化,流动相:A为0.1%TFA水溶液;流动相B为乙腈,梯度程序为:初始状态流动相B为10%,保持5分钟,然后在60分钟内将流动相B比例增至50%;流速为:80 mL/min。于226nm波长处检测,根据色谱峰分段收集馏分,并将收集的馏分用分析液相色谱仪进行检测,纯度≥99.0%且单杂≤0.2%馏分即为第一步合格馏分。其余为不合格馏分,不合格馏分中纯度大于80%部分再进行回收纯化,纯度小于80%舍弃。
(11)第二次纯化:将色谱柱用50%乙腈冲洗干净后用5%乙腈平衡系统5min,以20-35g/次的上样量进行HPLC纯化,流动相:A为0.1%醋酸水溶液;流动相B为乙腈,梯度程序为:初始状态流动相B为10%,保持5分钟,然后在60分钟内将流动相B比例增至50%,流速为:80 mL/min。于226nm波长处检测,根据色谱峰分段收集馏分,并将收集的馏分用分析液相色谱仪进行检测,纯度≥99.5%且单杂≤0.1%馏分即为第二步合格馏分。其余为不合格馏分,不合格馏分中纯度大于80%部分再进行回收纯化,纯度小于80%舍弃。
(12)转盐的条件:流 动 相:A相:50 mmol/L乙酸铵的水溶液:乙腈=95:5; B相:0.1 %醋酸的水溶液:乙腈=95:5;C相:0.1 %醋酸的水溶液:乙腈=50:50,梯度程序为:以流动相A等梯度洗脱25分钟, 转换成流动相B等梯度洗脱15分钟, 转换成流动相C等梯度洗脱40分钟。以20-35g/次的上样量进行转盐,按照上述梯度要求进行洗脱,于226nm波长处检测,将有紫外吸收值的样品收集合并。
(13)浓缩及冻干:转盐后合并馏分加入5%的醋酸,分十批分别旋蒸浓缩4h,至地加瑞克浓度为50g/l左右,每批旋蒸完后均放入冰箱于-20℃下冷冻保存,待冻干前,将十批浓缩液均置于20℃水浴复融4h后合并,冻干后得到醋酸地加瑞克精肽:286.1g,纯度:99.89%,收率:56.4%。醋酸地加瑞克的粘度为2.1mPas。
实施例3:
(1)向不锈钢溶液制备罐中加入3.528L的注射用水,
(2)加入220.5g的甘露醇,保持搅拌使物料溶解,
(3)加入88.2g的醋酸地加瑞克原料药,保持搅拌使物料溶解,
(4)加0.882L注射用水至全量。分别经过0.45μm和0.22μm微孔滤膜过滤,分装。冷冻干燥,加塞。
实施例4:
(1)向不锈钢溶液制备罐中加入5.120L的注射用水,
(2)加入160.0g的甘露醇,保持搅拌使物料溶解,
(3)加入128.0g的醋酸地加瑞克原料药,保持搅拌使物料溶解,
(4)加1.280L的注射用水至全量。分别经过0.45μm和0.22μm微孔滤膜过滤,分装。冷冻干燥,加塞。
实施例5:
采用BROOKFIELD DV-II型旋转黏度计,对于醋酸地加瑞克及注射用地加瑞克的黏度进行测量,方法如下所示:
(1)供试品的配置:分别取醋酸地加瑞克用纯净水溶解配制成20mg/ml的均一溶液或注射用地加瑞克用注射用水配制成20mg/ml的均一溶液。
(2)把盛有供试品的容器浸入20℃水浴。
(3)将转子浸入样品中至转子杆上的凹槽刻痕处。
(4)设定转速值为50rpm,开启搅拌检测供试品黏度,待仪器读数稳定后,记录数据。
其中实施例2中醋酸地加瑞克黏度值为:2.1mPas,实施例3中注射用地加瑞克黏度值为9.8mPas,实施例4中注射用地加瑞克黏度值为10.4mPas。
- 还没有人留言评论。精彩留言会获得点赞!