一种利用水稻SNB基因来增加穗粒数和降低株高的方法与流程
本发明涉及植物基因工程技术领域。具体涉及水稻SNB基因在增加穗粒数和降低株高中的应用。通过转基因的方法,在水稻中超量表达microRNA172结合位点突变的rSNB基因的蛋白质编码区显著的增加了穗粒数和降低了株高,表明SNB基因在水稻遗传改良中具有重要的利用价值。
背景技术:
随着全球人口的不断增长以及人类消费方式的改变,全球的粮食危机日益严峻。水稻是全球最重要的粮食作物之一,我国有超过一半的人口以稻谷为主粮,因此提高水稻的产量对保障国家的粮食安全具有重要的战略意义。
水稻的单株产量是由分蘖数、每穗颖花数和粒重共同决定的,其中每穗颖花数的多少取决于穗型的形成过程。水稻的穗是在进入生殖生长以后由花序分生组织分化而来的,在最终形成花器官之前,水稻的穗部会逐渐产生一次枝梗分生组织、二次枝梗分生组织和小穗分生组织,这些不同分生组织之间转变的时间在很大程度上决定了水稻的穗型(Kyozuka et al.,Control of grass inflorescence form by the fine-tuning of meristem phase change.Curr Opin Plant Biol,2014,17:110-115)。如果小穗分生组织的分化被推迟的话,则可能产生更多的二次枝梗甚至三次枝梗,从而可以提高最终的穗粒数。鉴于在农业生产上具有重要的利用价值,有关穗型的研究长期以来都是植物学研究的热门领域,科学家已经通过各种手段,分离克隆了多个参与穗型形成决定的基因,并且一些基因已经在育种实践中得到了应用(Zhang and Yuan,Molecular control of grass inflorescence development.Annu Rev Plant Biol,2014,65:553-578)。从目前分离到的基因来看,转录因子在其中占了很大的一部分,表明基因转录调控对穗型的形成具有重要的决定作用。申请人通过分析水稻多个连续发育阶段的幼穗的全基因组转录组也发现转录因子表达模式在不同的发育时期差别很大,这也进一步暗示转录因子在修饰穗型中发挥着核心的作用(Wang et al.,A dynamic gene expression atlas covering the entire life cycle of rice.Plant J,2010,61:752-766)。
株高是决定水稻产量的另外一个重要的因素。上个世纪以降低株高为目标的第一次“绿色革命”培育的半矮秆品种,极大的增加了全球农作物的产量。因为水稻株高和植株的倒伏性是密切相关的,株高矮化,可以提高作物的耐肥性,增加抗倒伏性,提高收获指数,从而可以大幅度增加产量。2002年水稻绿色革命基因SD1被成功克隆,其编码的是一种赤霉素氧化酶,控制赤霉素的合成(Sasaki et al.,Green revolution:a mutant gibberellins-synthesis gene in rice.Nature,2002,416:701-702)。除了赤霉素,油菜素甾醇、生长素、独脚金内酯和细胞分裂素等多种植物激素也参与决定植物的株高(Wang and Li,Molecular basis of plant architecture.Annu Rev Plant Biol,2008,59:253-279)。
MicroRNA172是一类在不同植物中高度保守的非编码小RNA,其通过调节AP2类的转录因子来发挥作用。AP2是最早被鉴定的参与花器官模式建成的A类基因,因此microRNA172和AP2之间的调控作用对花器官的形态建成具有重要的调节作用(Chen,A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development.Science,2004,303:2022-2025)。此外,受microRNA172调节的AP2类基因 还在植物多个发育过程中发挥作用,比如参与玉米的性别决定过程,也与大麦的闭花受精和密穗的形成过程密切相关(Chuck et al.,The maize tasselseed4microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1.Nat Genet,2007,39:1517-1521;Houston et al.,Variation in the interaction between alleles of HvAPETALA2and microRNA172determines the density of grains on the barley inflorescence.Proc Natl Acad Sci U S A,2013,110:16675-16680;Nair et al.,Cleistogamous flowering in barley arises from the suppression of microRNA-guided HvAP2mRNA cleavage.Proc Natl Acad Sci U S A,2010,107:490-495)。在水稻中已有两个受microRNA172调节的AP2类基因(SNB和OsIDS1)被报道参与花器官的发育,在它们的双突变体中,穗部分枝和颖花数会严重减少,因此这些基因的突变体等遗传材料无法直接用于水稻的育种(Lee and An,Two AP2family genes,SUPERNUMERARY BRACT(SNB)and OsINDETERMINATE SPIKELET 1(OsIDS1),synergistically control inflorescence architecture and floral meristem establishment in rice.Plant J,2012,69:445-461)。本发明通过超量表达microRNA172识别位点突变的rSNB基因的蛋白质编码区增加了水稻穗粒数和降低了水稻的株高,为在农业生产上利用SNB基因来对水稻进行遗传改良提供了新的方法。
技术实现要素:
本发明的目的在于提供一种SNB基因在增加水稻穗粒数和降低株高中的应用。本发明的另外一个目的是提供一种突变SNB基因中microRNA172结合位点的方法,以及超量表达microRNA172结合位点突变的rSNB基因的蛋白质编码区的载体和含有该载体的转基因水稻。同时本发明还提供了一种通过SNB及microRNA172结合位点突变的rSNB基因来增加水稻穗粒数和降低株高的方法。所述的SNB基因具有如SEQ ID NO:1所示的核苷酸序列,microRNA172结合位点突变的rSNB基因具有如SEQ ID NO:2所示的核苷酸序列,它们编码的蛋白质序列如SEQ ID NO:3所示。本发明也包括其他改变microRNA172结合位点而得到的rSNB的等位基因或衍生物。本发明通过超量表达microRNA172结合位点突变的rSNB基因的蛋白质编码区显著增加了水稻的穗粒数和降低了水稻的株高,这些结果表明SNB基因在水稻育种中可能具有重要的利用价值。SNB和microRNA172结合位点突变的rSNB基因可用于与其它调控元件,如组成型启动子或器官特异性启动子融合以构建基因表达载体转化水稻而改良重要的农艺性状。
实现本发明的具体技术方案如下:
1、分离水稻SNB基因的编码区的cDNA;具体方法在实施例1中有详细描述。
2、以上述cDNA作为模板,利用重叠延伸PCR的方法突变SNB基因中的microRNA172的识别位点;具体的方法在实施例2中有详细描述。
3、将microRNA172结合位点突变的rSNB基因的蛋白质编码区连接到pCAMBIA1301S载体上,构建超量表载体35S:rSNBOE;具体方法在实施例3中有详细描述。
4、利用优化过的农杆菌介导的转基因方法(Lin and Zhang,Optimising the tissue culture conditions forhigh efficiency transformation of indica rice.Plant Cell Rep,2005,23:540-548)将载体35S:rSNBOE导入水稻受体中花11(中国农业科学院作物科学研究所),获得转化植株;具体方法在实施例4中有详细描述。
5、借助PCR的方法分析鉴定阳性转基因植株和共分离检测,并对T1代单株进行表型鉴定和统计分析;具体方法在实施例5中有详细描述。
6、利用拟南芥原生质体瞬时表达系统,检测SNB蛋白质的转录活性;具体方法在实施例6中有详细 描述。
与现有技术相比,本发明的优点如下:
1、本发明克隆的SNB基因具有增加水稻穗粒数和降低株高的功能。
2、本发明将microRNA172识别位点突变的rSNB基因的蛋白质编码区导入水稻,实现了对水稻穗粒数和株高的遗传改良。
3、本发明建立了一种利用SNB基因来对水稻穗粒数和株高进行遗传改良的方法。
附图说明
序列表SEQ ID NO:1是SNB基因(原基因)的核苷酸序列。
序列表SEQ ID NO:2是microRNA172识别位点被人工同义突变的rSNB基因(突变后的基因)的核苷酸序列。
序列表SEQ ID NO:3是SNB基因和rSNB基因编码的蛋白质序列(虽然rSNB相对于SNB有核苷酸突变,但是它们编码的氨基酸是不变的,最大的区别就在于,突变后的rSNB基因与突变前的SNB基因相比活性有明显不同,这是本发明的显著特点)。
图1:SNB基因与microRNA172(有时简称miR172)的核苷酸配对示意图。附图标记说明:
图中红色字母表示SNB基因中microRNA172的结合位点突变后的序列,相对于SNB基因的序列,突变后形成的rSNB将不再受microRNA172调控,但是所编码的蛋白质依然与SNB基因是一样。
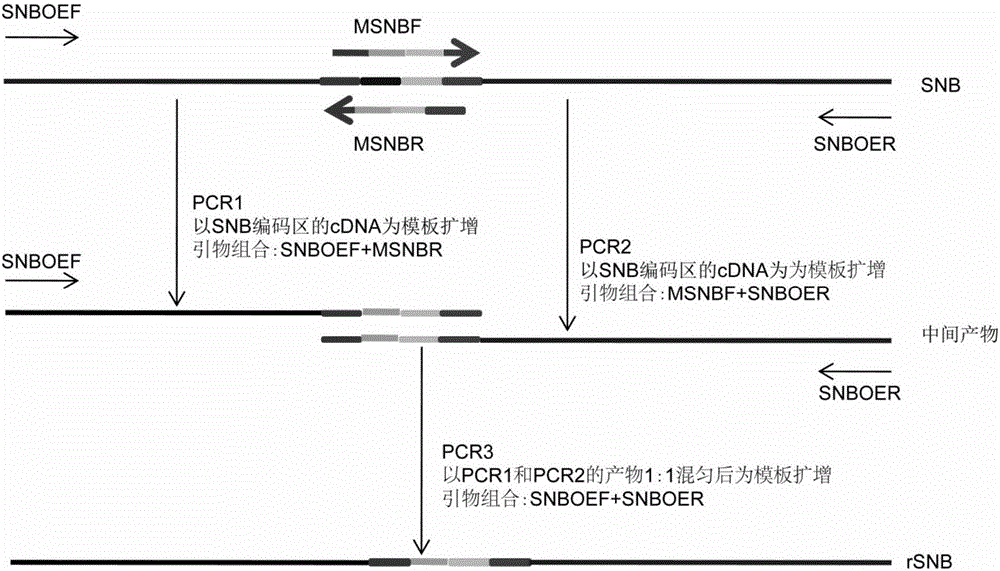
图2:利用重叠延伸PCR方法突变SNB基因中的microRNA172结合位点的示意图。
附图标记说明:红色、绿色和紫色片段代表SNB基因中用于设计引物的区段;蓝色片段代表SNB基因中的microRNA172的结合位点;橙色的片段代表SNB基因中的microRNA172的结合位点被突变后形成的序列。
图3:表达载体35S:rSNBOE构建过程示意图。附图标记说明:
图3中的A图:插入表达载体35S:rSNBOE的包含有rSNB蛋白质编码区的DNA片段示意图。
图3中的B图:用于构建表达载体35S:rSNBOE的空载体pCAMBIA1301S的结构示意图。
本发明将图3中的A图所示的片段通过BamHI和XbaI双酶切插入图3中的B图所示的载体pCAMBIA1301S的BamHI和XbaI酶切位点中间形成转化用的表达载体35S:rSNBOE。
图4:本发明构建的表达载体35S:rSNBOE结构示意图。
图5:35S:rSNBOE转基因T1代单株的基因型检测结果。附图标记说明:
图5中的A图:35S:rSNBOE转基因第一个家系的T1代植株基因型检测。所用的引物是SNBOEJCF和SNBOEJCR,612bp大小的产物是该引物对扩增的基因组DNA片段,196bp大小的产物是该引物对扩增的转入的rSNB基因的蛋白质编码区的片段。出现196bp大小的扩增产物的单株是转基因阳性单株。第一个泳道是DNA marker。
图5中的B图:35S:rSNBOE转基因第二个家系的T1代植株基因型检测。所用的引物是SNBOEJCF和SNBOEJCR,612bp大小的产物是该引物对扩增的基因组DNA片段,196bp大小的产物是该引物对扩增的转入的rSNB基因的蛋白质编码区的片段。出现196bp大小的扩增产物的单株是转基因阳性单株。第一个泳道是DNA marker。
图5中的C图:35S:rSNBOE转基因第三个家系的T1代植株基因型检测。所用的引物是SNBOEJCF 和SNBOEJCR,612bp大小的产物是该引物对扩增的基因组DNA片段,196bp大小的产物是该引物对扩增的转入的rSNB基因的蛋白质编码区的片段。出现196bp大小的扩增产物的单株是转基因阳性单株。第一个泳道是DNA marker。
图6:野生型中花11(WT)与35S:rSNBOE超量表达(rSNBOE)的植株的表型比较。附图标记说明:
图6中的A图:野生型中花11(WT)与35S:rSNBOE超量表达(rSNBOE)的植株的株高和分蘖的形态比较。
图6中的B图:野生型中花11(WT)与35S:rSNBOE超量表达(rSNBOE)的植株的穗型比较。
图6中的C图:野生型中花11(WT)与35S:rSNBOE超量表达(rSNBOE)的植株的一次枝梗形态比较。
图7:SNB基因的转录活性检测所用的载体示意图。附图标记说明:
图7中的A图:用于SNB基因转录活性检测的效应子对照35S:GAL4的结构示意图。
图7中的B图:用于SNB基因转录活性检测的效应子35S:GAL4-SNB的结构示意图。
图7中的C图:用于SNB基因转录活性检测的报告子4×UAS:mini35S:LUC的结构示意图。
图7中的D图:用于SNB基因转录活性检测的内参35S:GUS的结构示意图。
图8:SNB基因的转录活性检测的结果。
将报告子、效应子和内参的质粒按照5:4:1的比例通过聚乙二醇介导共转化进入拟南芥原生质体,并于24h后测量酶活。利用内参的GUS活性来平衡化各个转化之间的系统误差,并将与效应子GAL4共转化的荧光素酶酶活设置为相对活性1。
具体实施方式
实施例1:水稻SNB基因的克隆
本发明采用水稻叶片的RNA经过反转录得到的cDNA为模板,利用SNB基因(LOC_Os07g13170,其核苷酸序列如SEQ ID NO:1所示)的特异引物,通过PCR的方法分离克隆SNB基因的蛋白质编码区,具体的操作如下:
(1)提取水稻叶片RNA
抽提水稻品种中花11(中国农业科学院作物科学研究所)植株分蘖期叶片的RNA,RNA抽提用的试剂购采用Invitrogen公司生产的Trizol抽提试剂盒(具体操作步骤按照试剂盒提供的说明书操作)。
(2)反转录合成cDNA第一链
步骤如下:1)取抽提的总RNA 2μg,加入DNaseI 1μl,10xDNaseI buffer 1μl,加DEPC(焦碳酸二乙酯,RNA酶的强烈抑制剂,工作浓度为0.01%)处理过的水到10μl,混匀后于室温放置15min以去除残留的基因组DNA;2)15min后加入0.2M EDTA 1μl,并于65℃水浴中孵育10min以去除DNaseI的活性;3)加入1μl引物oligo(dT)15,并于65℃水浴中孵育10min以破坏RNA的二级结构,然后冰上放置5min;4)加入5x first strand buffer 4μl,0.1M DTT(巯基乙醇)2μl,10mM dNTP mixture 1μl,反转录酶1μl,混匀后置于42℃水浴锅内温浴1.5h;5)反应结束后将反转录产物置于90℃干浴3min以灭活反转录酶;6)向反转录产物中加入120μl水,混匀后-20℃保存反应最终产物。反应中用到的试剂全部购自Invitrogen公司。
(3)扩增SNB基因的编码区的mRNA
为了扩增SNB基因编码区的mRNA,本发明设计如下引物:
SNBOEF(正向引物):5'-GGATCCATGGTGCTGGATCTCAATGTGG-3'(下划线序列为BamHI识别位点);
SNBOER(反向引物):5'-TCTAGATTAGGCGGTCGGGGGGAAGTAGA-3'(下划线序列为XbaI识别位点);
该引物对可以扩增出SNB基因的起始密码子到终止密码子之间的蛋白质编码区段。
以步骤(2)反转录而来的cDNA为模板,用引物SNBOEF和SNBOER进行PCR扩增。PCR反应总体积为20μl,包含cDNA模板1μl,10xPCR buffer 2μl,10mM dNTP 2μl,10mM引物SNBOEF和SNBOER各0.3μl,ExTaq酶0.2μl,加去离子水至20μl(所用到的PCR buffer、dNTP、rTaq酶等均购自宝生物工程大连有限公司)。PCR反应条件如下:①94℃4min,②94℃40s,③58℃40s,④72℃2min,⑤从②-④循环38次,⑥72℃7min,⑦4℃保存。PCR产物在1%(质量/体积)的TBE琼脂糖凝胶上电泳检测,回收长度为1323bp(1311bp的目标DNA区段加上引物上附加的两个限制性酶切位点12bp)的DNA片段。
实施例2:SNB中microRNA172结合位点的突变
因为SNB受到microRNA172的调控(如图1所示),因此为了超量表达SNB,本发明采用重叠延伸PCR的方法突变掉SNB基因中的microRNA172的结合位点(重叠延伸PCR方法参考萨姆布鲁克,《分子克隆实验指南》第三版,科学出版社,2002年;基本流程如图2所示)。SNB中microRNA172的结合位点序列为TGCAGCATCATCAGGATTCT,本发明将该结合位点突变为CGCCGCCAGCAGCGGCTTTT,突变后的SNB基因命名为rSNB基因。rSNB基因将不再受microRNA172调控,但是该突变是同义突变,因此并不改变SNB编码的蛋白质的序列。
为实现以上目标,设计以下引物:
MSNBF(正向引物):5'-CCTACCGCCGCCAGCAGCGGCTTTTCTACGGGCACCGTCGCC-3'(下划线序列为microRNA172结合位点被突变以后的序列);
MSNBR(反向引物):5'-CCGTAGAAAAGCCGCTGCTGGCGGCGGTAGGGAGTAATGGCA-3'(下划线序列为microRNA172结合位点被突变以后的互补序列)。
以实施例1中所得PCR产物为模板再进行PCR。分别以引物SNBOEF和MSNBR,MSNBF和SNBOER为组合进行两组PCR,PCR体系及程序与实施例1中所述的PCR体系及程序一致。从以上2组PCR反应产物中各取0.5μl混合后作为下一轮重叠延伸PCR的模板,以引物SNBOEF和SNBOER为组合进行PCR扩增,PCR体系及程序与实施例1中所述的PCR体系及程序一致。最终得到的PCR产物就是microRNA172结合位点被突变掉的rSNB基因的蛋白质编码区,其序列如SEQ ID NO:2所示,所编码的蛋白质的序列如SEQ ID NO:3所示。将该PCR产物在1%(质量/体积)的TBE琼脂糖凝胶上电泳检测,并将回收的PCR产物连接到T-A克隆载体pGEMT-vector上(购自普洛麦格(北京)生物技术有限公司,即美国Promega公司),利用载体上T7和SP6引物对该PCR产物进行测序验证。将包含该PCR产物的重组质粒命名为质粒TA-rSNB。
实施例3:microRNA172识别位点突变的rSNB基因的蛋白质编码区的超量表达载体构建
用BamHI和XbaI双酶切质粒TA-rSNB(所述的BamHI和XbaI购买自宝生物工程大连有限公司,具 体用法与用量参考该公司相应产品的说明书,质粒TA-rSNB来自实施例2,如图3中的A图所示),回收包含rSNB的1323bp的DNA片段,然后与经过BamHI和XbaI双酶切的载体质粒pCAMBIA1301S[该载体被公开报道:Zhou et al.,Over-expression of aspartate aminotransferase genes in rice resulted in altered nitrogen metabolism and increased amino acid content in seeds.Theor Appl Genet,2009,118:1381-1390;其基本骨架起源于澳大利亚的CAMBIA实验室(http://www.cambia.org/daisy/cambia/materials/overview.html)的pCAMBIA1301,通过加入35S启动子实现对转化基因的表达调控,其结构如图3中的B图所示]利用T4DNA连接酶(购自Promega公司,具体用法与用量参考该公司产品的说明书)进行连接。连接产物通过电转化的方法(电转化仪为eppendorf公司产品,本发明所用电压为1800V,具体操作参考该仪器的使用说明书)导入大肠杆菌DH10B(购自Promega公司)中,加400μl LB培养基复苏45min,涂于含50mg/L的卡那霉素的LA培养基平板上,37℃温箱培养14-16h(LA与LB配方参考:萨姆布鲁克,《分子克隆实验指南》第三版,科学出版社,2002年)。挑取单克隆,扩大培养并抽提质粒,通过BamHI和XbaI双酶切筛选阳性克隆,并将所得的表达载体命名为35S:rSNBOE,其结构如图4所示。
实施例4:转基因水稻的获得
将表达载体35S:rSNBOE(来自实施例3)通过农杆菌EHA105介导的遗传转化方法导入水稻品种中花11的愈伤组织,经过预培养、侵染、共培养、筛选具有潮霉素(用于筛选阳性的转基因愈伤的一种抗生素,购买自丹麦的罗氏制药有限公司)抗性的愈伤组织、分化、生根及炼苗移栽大田,得到转基因植株。农杆菌遗传转化的方法和所用的试剂及配方是根据Hiei等人的报道优化而来(Hiei et al.,Efficient transformation of rice(Oryza sativa L.)mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA.Plant J,1994,6:271-282;Lin and Zhang,Optimising the tissue culture conditions for high efficiency transformation of indica rice.Plant Cell Rep,2005,23:540-548)。
本发明所涉及到的农杆菌介导的遗传转化试剂及配方如下:
(1)试剂和溶液缩写
6-BA(6-BenzylaminoPurine,6-苄基腺嘌呤);KT(Kinetin,激动素);NAA(Napthalene acetic acid,萘乙酸);IAA(Indole-3-acetic acid,吲哚乙酸);2,4-D(2,4-Dichlorophenoxyacetic acid,2,4-二氯苯氧乙酸);AS(Acetosringone,乙酰丁香酮);CH(Casein Enzymatic Hydrolysate,水解酪蛋白);HN(Hygromycin B,潮霉素);DMSO(Dimethyl Sulfoxide,二甲基亚砜);N6max(N6大量成分溶液);N6min(N6小量成分溶液);MSmax(MS大量成分溶液);MSmin(MS小量成分溶液)
(2)主要溶液配方
1)N6max母液[10倍浓缩液(10X)]
逐一溶解,然后室温下定容至1000ml。
2)N6min母液[100倍浓缩液(100X)]
室温下溶解并定容至1000ml。
3)Fe2-EDTA贮存液(100X)
在一个大三角瓶中加入300ml蒸馏水和硫酸铁(FeSO4·7H2O)2.78g
在另一个大三角瓶中加入300ml蒸馏水并加热至70℃,然后加入乙二铵四乙酸二钠(Na2EDTA·2H2O)3.73g
在它们都溶解后混合在一起,70℃水浴中保持2h,定容至1000ml,4℃保存备用。
4)维生素贮存液(100X)
加水定容至1000ml,4℃保存备用。
5)MSmax母液(10X)
室温下溶解并定容至1000ml。
6)MSmin母液(100X)
室温下溶解并定容至1000ml。
7)2,4-D贮存液(1mg/ml)
2,4-D 100mg.
1ml 1N氢氧化钾溶解5min,然后加10ml蒸馏水溶解完全后定容至100ml,室温保存。
8)6-BA贮存液(1mg/ml)
6-BA 100mg.
1ml 1N氢氧化钾溶解5min,然后加10ml蒸馏水溶解完全后定容至100ml,室温保存。
9)NAA贮存液(1mg/ml)
NAA 100mg.
1ml 1N氢氧化钾溶解5min,然后加10ml蒸馏水溶解完全后定容至100ml,4℃保存备用。
10)IAA贮存液(1mg/ml)
IAA 100mg.
1ml 1N氢氧化钾溶解5min,然后加10ml蒸馏水溶解完全后定容至100ml,4℃保存备用。
11)葡萄糖贮存液(0.5g/ml)
葡萄糖 125g
蒸馏水溶解定容至250ml,灭菌后4℃保存备用。
12)AS贮存液
AS 0.392g
DMSO 10ml
分装至1.5ml离心管内,4℃保存备用。
13)1N氢氧化钾贮存液
氢氧化钾(KOH)5.6g
蒸馏水溶解定容至100ml,室温保存备用。
14)KT贮存液(1mg/ml)
KT 100mg.
1ml 1N氢氧化钾溶解5min,然后加10ml蒸馏水溶解完全后定容至100ml,室温保存。
(3)培养基配方
1)诱导培养基
加蒸馏水至900ml,1N氢氧化钾调节pH值到5.9,煮沸并定容至1000ml,分装到50ml三角瓶(25ml/瓶),封口灭菌。
2)继代培养基
加蒸馏水至900ml,1N氢氧化钾调节pH值到5.9,煮沸并定容至1000ml,分装到50ml三角瓶(25ml/瓶),封口灭菌。
3)预培养基
加蒸馏水至250ml,1N氢氧化钾调节pH值到5.6,封口灭菌。
使用前加热溶解培养基并加入5ml葡萄糖贮存液和250μl AS贮存液,分装倒入培养皿中(25ml/皿)。
4)共培养基
加蒸馏水至250ml,1N氢氧化钾调节pH值到5.6,封口灭菌。
使用前加热溶解培养基并加入5ml葡萄糖贮存液和250μl AS贮存液,分装倒入培养皿中(25ml/皿)。
5)悬浮培养基
加蒸馏水至100ml,调节pH值到5.4,分装到两个100ml的三角瓶中,封口灭菌。
使用前加入1ml葡萄糖贮存液和100μlAS贮存液。
6)选择培养基
加蒸馏水至250ml,调节pH值到6.0,封口灭菌。
使用前溶解培养基,加入250μl 50mg/ml的潮霉素和400ppm头孢霉素,分装倒入培养皿中(25ml/皿)。
7)预分化培养基
加蒸馏水至250ml,1N氢氧化钾调节pH值到5.9,封口灭菌。
使用前溶解培养基,加入250μl 50mg/ml的潮霉素和400ppm头孢霉素,分装倒入培养皿中(25ml/皿)。
8)分化培养基
加蒸馏水至900ml,1N氢氧化钾调节pH值到6.0。
煮沸并定容至1000ml,分装到100ml三角瓶(50ml/瓶),封口灭菌。
9)生根培养基
加蒸馏水至900ml,1N氢氧化钾调节pH值到5.8。
煮沸并定容至1000ml,分装到生根管中(25ml/管),封口灭菌。
10)LA培养基(LB培养基不含琼脂粉)
蒸馏水溶解定容至250ml,装于500ml三角瓶,灭菌后室温保存备用。
本发明所涉及到的农杆菌介导的遗传转化的步骤如下:
(1)愈伤诱导
1)将成熟的水稻种子去壳,然后依次用70%的乙醇处理1min,0.15%氯化汞(HgCl2)15min
2)灭菌水洗种子4-5次;
3)将种子放在诱导培养基上;
4)置于黑暗处培养5周,温度26±1℃。
(2)愈伤继代
挑选亮黄色、紧实且相对干燥的胚性愈伤,放于继代培养基上黑暗下培养2周,温度26±1℃。
(3)预培养
挑选紧实且相对干燥的胚性愈伤,放于预培养基上黑暗下培养4d,温度26±1℃。
(4)农杆菌培养
1)在带有卡那霉素的LA培养基上预培养含构建好载体的农杆菌EHA1052d,温度28℃;
2)将农杆菌转移至悬浮培养基里,28℃摇床上培养2-3h。
(5)农杆菌侵染
1)将预培养的愈伤转移至灭菌好的瓶子内;
2)调节农杆菌的悬浮液至OD6000.8-1.0;
3)将愈伤在农杆菌悬浮液中浸泡30min;
4)转移愈伤至灭菌好的滤纸上吸干;然后放置在共培养基上培养2d,温度19-20℃。
(6)愈伤洗涤和选择培养
1)灭菌水洗涤愈伤至看不见农杆菌;
2)浸泡在含400ppm头孢霉素的灭菌水中30min;
3)转移愈伤至灭菌好的滤纸上吸干;
4)转移愈伤至选择培养基上选择2-3次,每次2周。(第一次头孢霉素筛选浓度为400ppm,第二次以后为250ppm)
(7)分化
1)将抗性愈伤转移至预分化培养基上黑暗处培养5-7d;
2)转移预分化培养的愈伤至分化培养基上,光照下培养,温度26℃,5-7周。
(8)生根
1)拔出分化好的苗子,剪掉分化时产生的根;
2)然后将其转移至生根培养基中光照下培养2-3周,温度26℃。
(9)移栽
洗掉根上的残留培养基,将具有良好根系的幼苗转入温室,同时在最初的几天保持水分湿润。在温室炼苗约2周左右后,再移载至大田。
实施例5:转基因水稻的鉴定与表型观察
设计以下引物用于检测转基因植株的基因型,序列如下:
SNBOEJCF(正向引物):5'-GCGATGGGAGTCGCACATCT-3';
SNBOEJCR(反向引物):5'-CACAAACTCCTCCTTGGTCCA-3';
因为SNBOEJCF是设计在SNB基因的第一个外显子上的,而SNBOEJCR是设计在SNB基因的第五个外显子上的,而遗传转化用的是没有内含子的突变了microRNA172结合位点的rSNB基因的蛋白质编码区进行的,因此使用该对引物扩增转基因植株,可以得到一条来自基因组DNA的612bp的片段,同时如果是转基因阳性植株的话,还可以得到一条来自rSNB基因蛋白质编码区的191bp的片段。根据这两条带型就可以判断PCR是否成功以及所检测的单株是否是转基因阳性的。
抽提T0代35S:rSNBOE转化植株(来自实施例4)叶片的总DNA,DNA抽提方法为CTAB法(Zhang et al.,Genetic diversity and differentiation of indica an japonica rice detected by RFLP analysis.Theor Appl Genet,1992,83:495-499)。以叶片总DNA为模板,用引物SNBOEJCF和SNBOEJCR进行PCR扩增。PCR反应总体积为20μl,包含DNA模板100ng,10xPCR buffer 2μl,10mM dNTP 2μl,10mM引物SNBOEJCF和SNBOEJCR各0.3μl,rTaq酶0.2μl,加去离子水至20μl(所用到的PCR buffer、dNTP、rTaq酶等均购自宝生物工程大连有限公司)。PCR反应条件如下:①94℃4min,②94℃40s,③58℃40s,④72℃1min,⑤从②-④循环38次,⑥72℃7min,⑦4℃保存。PCR产物在1%(质量/体积)的TBE琼脂糖凝胶上电泳检测。
PCR检测结果表明,对表达载体35S:rSNBOE转基因而来阳性植株相对于阴性植株而言,全部表现为株高变矮,每穗一次枝梗数不变,但是每穗二次枝梗数和每穗粒数都明显增加,其表型如图6所示。
为了进行表型分析,申请人将35S:rSNBOE的3个独立转化而来的T0植株的种子(来自实施例4)经过浸种、催芽后播种于秧田,20d以后移栽至大田获得T1代转基因植株。种植密度为15cm×24cm,种植地点为中国湖北省武汉市洪山区华中农业大学的试验田,在一个有安全防护设施条件下按常规的水稻种植方法进行田间管理。利用引物SNBOEJCF和SNBOEJCR进行PCR扩增以检测分离单株的阴阳性,并且进行转基因事件和表型变化的共分离检测,以及统计表型数据。
分析结果表明在T1代,表达载体35S:rSNBOE转基因后代分离出来的所有阳性单株相对于阴性单株而言,都表现为株高变矮,每穗一次枝梗数不变,但是每穗二次枝梗数和每穗粒数都明显增加,表明35S:rSNBOE的转基因事件同这些表型的变化是共分离的(见图5和图6所示的结果)。
考察和记录T1代相关植株的表型数据,包括株高、每穗一次枝梗数、每穗二次枝梗数和每穗粒数,并以每个家系分离出来的的阴性单株为对照,进行统计学分析。
表1超量表达35S:rSNBOE(rSNBOE)的T1代阳性和阴性单株的表型统计值
表1的说明:(+)和(-)分别表示转基因阳性和阴性单株。利用t测验进行统计学分析,a,b,c分别表示P<0.05,0.01和0.001的显著水平。
实施例6:SNB的转录活性检测
为了检测SNB的转录活性,申请人按照公开发表过的检测植物转录因子转录活性的方法进行(Weng et al.,Grain number,plant height,and heading date7is a central regulator of growth,development,and stress response.Plant Physiol,2014,164:735-747)。具体的讲,将SNB蛋白质融合到GAL4蛋白质(登陆号NM_001184062.1)的DNA结合结构域后面,并且将该重组蛋白质置于35S启动子控制下,构成检测转录活性的效应子(如图7中的A和B图所示)。GAL4蛋白质的DNA结合结构域识别的DNA元件称为UAS序列,申请人将4×UAS和mini35S的启动子序列克隆到报告基因萤火虫的荧光素酶前面构成效应子(如图7中的C图所示)。同时申请人还构建了一个35S启动子驱动的GUS基因(缩写为35S:GUS)作为不同转化之间的平衡化参照(如图7中D图所示)。申请人将报告子、效应子和参照35S:GUS的质粒按照5:4:1的比例,通过聚乙二醇介导的方法共转化进入拟南芥的原生质体,经过24h的室温培养,提取蛋白质测量荧光素酶的活性。聚乙二醇介导的拟南芥原生质体转化按照Yoo等报道的方法进行(Yoo et al.,Arabidopsis mesophyll protoplasts:a versatile cell system for transient gene expression analysis.Nat Protoc,2007,2:1565-1572);荧光素酶酶活测定的底物荧光素购买自普洛麦格(北京)生物技术有限公司,即美国Promega公司,具体的操作参照该公司的相关产品说明书进行;荧光素酶酶活测定所使用的仪器是TECAN infinite M200型多功能酶标仪,具体操作按照仪器的使用说明书进行。荧光素酶酶活检测结果见图8,相对于单独的GAL4的DNA结合结构域而言,融合有SNB蛋白质的GAL4的DNA结合结构域会导致报告基因荧光素酶的活性下降,因此SNB蛋白质在拟南芥原生质体中具有转录抑制子活性。
- 还没有人留言评论。精彩留言会获得点赞!