芳基胍类化合物及其制备方法和用途与流程
本发明涉及药物化学和药物治疗学领域,更具体涉及取代的芳基胍类化合物及其制备方法、药物组合物和用途。该类化合物可用于制备治疗肿瘤的药物。
背景技术:
恶性肿瘤作为全球较大的公共卫生问题之一,极大地危害人类的健康,并将成为新世纪人类的第一杀手。恶性肿瘤已不再只是发达工业国家的严重疾病,发展中国家面临着更大的疾病负担。
多发性骨髓瘤(multiplemyeloma,mm)是一种克隆性浆细胞异常增殖的恶性疾病,是血液系统第二位常见的恶性肿瘤,约占血液系统恶性肿瘤的10%,多发于中老年人群,目前仍无法治愈,其中位生存时间为5~6年。传统治疗多发性骨髓瘤的主要方法是化疗和造血干细胞移植,其临床疗效很难维持。近10年来,随着新型药物如蛋白酶体抑制剂硼替佐米、免疫调节剂沙利度胺及来那度胺等的出现,多发性骨髓瘤患者的完全缓解率及总体生存率明显提高。但同时仍存在以下不足:首先,以上这些药物在复发/难治患者中的单药有效率仅为25%~50%;其次,尽管无疾病生存时间得到延长,但多数患者终将复发,且出现显著的药物抵抗;第三,神经炎等某些严重的副作用限制了药物的应用。因而,研发、检验新的治疗药物仍然是目前多发性骨髓瘤治疗所需面临的重要难题。
淋巴瘤是血液系统最常见的恶性肿瘤之一,在我国常见恶性肿瘤中占第8位,并且近年来它的发病率仍在逐渐增加。新的化疗方案、单克隆抗体、细胞免疫治疗等治疗方式已经显著改善了淋巴瘤患者的生存。特别是利妥昔单抗的出现,淋巴瘤的治疗有了突破性的进展,尤其是对cd20阳性的b细胞淋巴瘤,有效率更高,缓解时间更长,显著改善预后。但是,淋巴瘤患者出现复发或耐药的比例仍然很高。因此仍需进一步研发新药,以提高淋巴瘤的治疗效果与治愈率。
虽然化学药物治疗作为治疗肿瘤的重要手段之一,在近三十年已经有了巨大的发展和进步,得到了一大批具有不同作用机制的临床抗肿瘤药物。但是抗肿瘤药也存在许多不良反应,比如脱发,呕吐,快速产生耐药性等等,这些都导致化学药物无法达到预期的治疗效果。因此,新的抗肿瘤药物的研究与开发是目前药学领域的热点和难点问题之一。
索拉菲尼是一种临床应用广泛的广谱抗肿瘤药物,具有抗肾癌、肝癌、胃癌等作用。
胍基官能团存在于很多药物分子中,例如大名鼎鼎的抗糖尿病药物二甲双胍,其 分子中的胍基质子化后带正电荷,能与蛋白质中的胍基、羧基等基团发生相互作用,产生意想不到的生物效应。
本发明将索拉菲尼的活性脲羰基变换成胍基后合成出了一系列结构新颖的化合物,结果发现,与索拉菲尼相比,本发明的化合物针对肿瘤细胞的活性有较大改变,总的来说抗肿瘤活性选择性更好,尤其对骨髓瘤和淋巴瘤细胞有较好的抑制效果,因此,具有开发成预防或治疗该类疾病药物的潜在价值。
技术实现要素:
本发明的目的在于提供一种芳基胍类化合物及其制备方法和应用,该化合物在体外表现出很好的抗肿瘤活性,能够应用于抗肿瘤药物的制备。
本发明的第一方面,
提供一种通式i所示的化合物或其药学上可接受的盐:
其中:
q为
r1、r2各自独立的选自未取代或取代的c6-c20芳基或c3-c14杂芳基,其中所述杂芳基具有一个或多个选自下组的杂原子:o、n或s;
l为未取代或取代的-(ch2)n-、或未取代或取代的
z为氢、烷基、未取代或取代的c6-c20芳基或c3-c14杂芳基,其中所述杂芳基具有一个或多个选自下组的杂原子:o、n或s;
r1、r2、l和z中各所述取代包括但不限于被选自如下的一种或多种取代基所取代:卤素、羟基、c1-c6烷基、c3-c8环烷基、c1-c6烷氧基、氨基、-nh(c1-c6烷基)、-n(c1-c6烷基)(c1-c6烷基)、硝基、c2-c6烯基、卤代c1-c6烷基、羰基、羧基、酰胺基、氰基、羟甲基、卤代c1-c6烷氧基、巯基、氨磺酰基、苯基、苯氧基;
所述芳基包括但不限于:苯基、萘基、四氢萘基、蒽基、菲基、芘基;
所述杂芳基包括但不限于:吡啶基、苯并噻唑基、呋喃基、芴基、吡咯基、噻吩基、噁唑基、咪唑基、噻唑基、嘧啶基、喹唑啉基、喹啉基、异喹啉基、吲哚基。
优选的,
r1、r2各自独立的选自未取代或取代的c6芳基或c5-c6杂芳基,其中所述杂芳基具有一个或多个选自下组的杂原子:o、n或s;所述取代是指被卤素或酰胺基所取代;
l为
z为叔丁基、未取代或取代的c6芳基或未取代或取代的c5-c6杂芳基,其中所述杂芳基具有一个或多个选自下组的杂原子:o、n或s;所述取代是指被选自如下的一种或多种取代基所取代:卤素、c1-c6烷基、c1-c6烷氧基、卤代c1-c6烷基。
更优选的,
r1、r2各自独立的选自未取代或取代的c6芳基或c5-c6杂芳基,其中所述杂芳基具有一个或多个选自下组的杂原子:o、n或s;所述取代是指被卤素或酰胺基所取代;
l为
z为未取代或取代的c6芳基或未取代或取代的c5-c6杂芳基,其中所述杂芳基具有一个或多个选自下组的杂原子:o、n或s;所述取代是指被选自如下的一种或多种取代基所取代:卤素、c1-c6烷氧基、卤代c1-c6烷基。
进一步优选的,所述通式i所示的化合物选自:
通过体外酶活性抑制试验,证实了本申请化合物具有较强的酶活性抑制能力,并且进一步的细胞水平检测,发现本发明化合物具有明显的抗肿瘤活性,可发展为肿瘤预防和/或治疗药物。
本发明的第二方面,
提供通式i所示的化合物的制备方法,所述方法包括以下步骤:
式a化合物溶于有机溶剂,加入缩合剂或者氧化剂,与含氮的亲核试剂反应,生成含式i所示化合物的混合物,然后通过常规的分离纯化技术获得式i化合物。
其中z、l和q的定义如前所述。
所述有机溶剂为n,n-二甲基甲酰胺、n-甲基吡咯烷酮、n,n-二甲基乙酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、三氯甲烷中的一种或两种以上的混合物;
所述氧化剂为2-碘酰基苯甲酸、碘单质、氧气、双氧水、二氧化锰、重铬酸钾、硝酸铈铵中的一种或两种以上的混合物;
所述缩合剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、二环己基碳二亚胺、n,n'-二异丙基碳二亚胺、1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐中的一种或两种以上的混合物;
所述含氮的亲核试剂为氨水、液氨、氨甲醇、氨气、六甲基二硅氮烷中的一种或两种以上的混合物;
本发明化合物可以任选将在本说明书中描述的或本领域已知的各种合成方法组合起来而方便的制得。
本发明的第三方面,
提供一种药物组合物,包含:(1)通式i所示的化合物或其药学上可接受的盐;和(2)药学上可接受的载体。
本发明的第四方面,
提供通式i所示的化合物或其药学上可接受的盐的用途,用于制备预防和/或治疗肿瘤的药物。
所述相关的肿瘤选自:口咽癌、结直肠癌、非小细胞肺癌、神经胶质瘤、白血病、肝癌、甲状腺癌、食道癌、淋巴瘤、胸腔癌、消化道癌、胰腺癌、乳腺癌、卵巢癌、子宫癌、肾癌、胆囊癌、胆管癌、中枢神经癌、睾丸癌、膀胱癌、前列腺癌、皮肤癌、黑色素瘤、肉癌、脑癌、宫颈癌、胶质瘤、胃癌、或腹水瘤、骨髓瘤。
本发明的第五方面,
提供一种体外非治疗性的抑制肿瘤细胞的增殖或诱导肿瘤细胞的凋亡的方法,将通式i所示的化合物或其药学上可接受的盐或所述的其药物组合物与所述肿瘤细胞接触,从而抑制肿瘤细胞的增殖或诱导肿瘤细胞的凋亡。
本发明发现了一类新型的取代的芳基胍类化合物,通过体外酶活性抑制试验,证 实了本申请化合物具有较强的酶活性抑制能力,并且进一步的细胞水平检测,发现本发明化合物具有明显的抗肿瘤活性,可发展为肿瘤预防和/或治疗药物。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
图1为20天后处死小鼠,取出肿瘤的肿瘤照片;
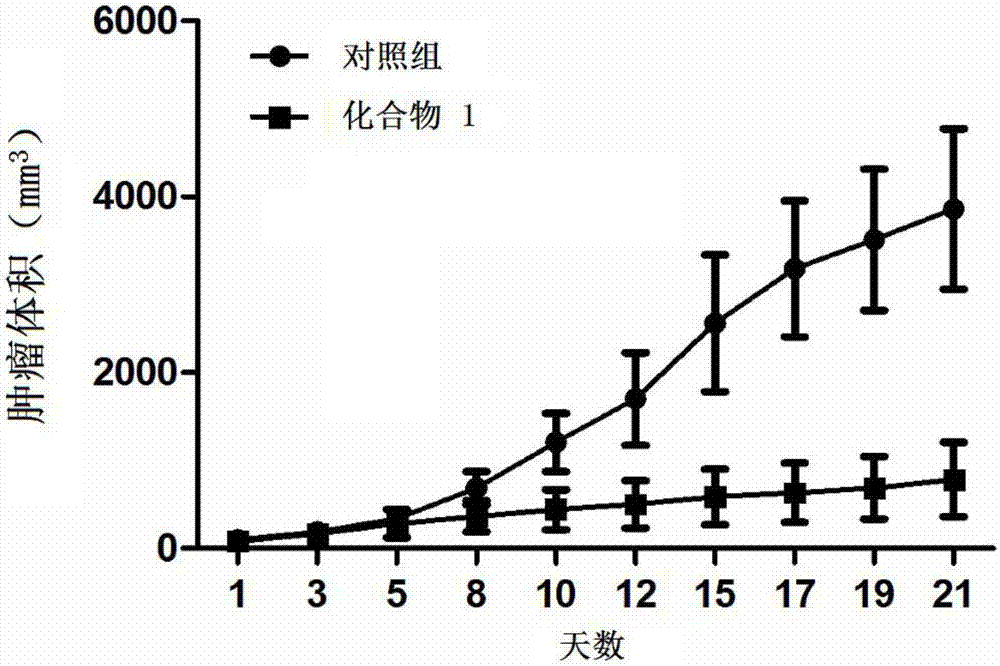
图2为小鼠肿瘤体积线形图。
具体实施方式
在以下的实施例中将进一步举例说明本发明。这些实施例仅用于说明本发明,但不以任何方式限制本发明。实施例中的所有参数以及其余的说明,除另有说明外,都是以质量(克)为单位。
术语
本发明的上下文中,术语“烷基”表示饱和的线性或支链烃部分,例如-ch3或-ch(ch3)2。术语“烷氧基”表示-o-(c1-6烷基)基团。术语“烯基”表示包含至少一个双键的直链或支链烃基部分,例如-ch=ch-ch3。术语“环烷基”表示饱和的环状烃基部分,例如环己基。术语“芳基”表示包含一个或多个芳环的烃基部分。芳基部分的例子包括但不限于苯基(ph)、萘基、芘基、蒽基和菲基。术语“杂芳基”表示包含一个或多个具有至少一个杂原子(例如n,o或s)的芳环的部分。杂芳基部分的例子包括呋喃基、芴基、吡咯基、噻吩基、噁唑基、咪唑基、噻唑基、吡啶基、嘧啶基、喹唑啉基、喹啉基、异喹啉基和吲哚基。
除非另外说明,本文所述的烷基、烯基、环烷基、芳基和杂芳基同时包括取代的和未取代的部分。烷基、烯基、环烷基、芳基和杂芳基上可能的取代基包括,但不限于:c1-c10烷基、c2-c10烯基、c2-c10炔基、c3-c20环烷基、c3-c20环烯基、c1-c20杂环烷基、c1-c20杂环烯基、c1-c10烷氧基、芳基、杂芳基、杂芳氧基、氨基、c1-c10烷基氨基、c1-c20二烷基氨基、芳基氨基、二芳基氨基、c1-c10烷基氨磺酰基、芳基氨磺酰基、c1-c10烷基亚氨基、c1-c10烷基磺基亚氨基、芳基磺基亚氨基、羟基、卤素、巯基、c1-c10烷硫基、c1-c10烷基磺酰基、芳基磺酰基、酰基氨基、氨酰基、氨基硫代酰基、胍基、脲基、氰基、硝基、酰基、硫代酰基、酰氧基、 羧基和羧酸酯基。另一方面,环烷基、杂环烷基、杂环烯基、芳基和杂芳基也可互相稠合。
本发明中,所述取代为单取代或多取代,所述多取代为二取代、三取代、四取代、或五取代。所述二取代就是指具有两个取代基,依此类推。
本发明所述药学上可接受的盐可以是阴离子与式i化合物上带正电荷的基团形成的盐。合适的阴离子包括氯离子、溴离子、碘离子、硫酸根、硝酸根、磷酸根、柠檬酸根、甲基磺酸根、三氟乙酸根、乙酸根、苹果酸根、甲苯磺酸根、酒石酸根、富马酸根、谷氨酸根、葡糖醛酸根、乳酸根、戊二酸根和马来酸根。类似地,可以由阳离子与式i化合物上的带负电荷的基团(例如羧酸根)形成盐。合适的阳离子包括钠离子、钾离子、镁离子、钙离子和铵离子,例如四甲基铵离子。
在另一优选例中,“药学上可接受的盐”是指同选自下述酸形成的盐类:氢氟酸、盐酸、氢溴酸、磷酸、乙酸、草酸、硫酸、甲磺酸、水杨酸、三氟甲磺酸、萘磺酸、马来酸、柠檬酸、醋酸、酒石酸、琥珀酸、酢浆草酸、苹果酸、谷氨酸。
制备实施例:
薄层分析(tlc)板型号为hsgf-254(厚度0.15-0.2mm,烟台化工实验厂生产);柱层析硅胶为青岛海洋化工厂生产200-300目商业化硅胶;1h-nmr,13c-nmr使用brukeram-300、varianmercury-400或者varianmercury-500核磁共振仪记录,内标为四甲基硅烷(tms);化学位移为(ppm,δ),质子偶合标记为单重峰(s),双重峰(d),三重峰(t),四重峰(q),多重峰(m),宽峰(br);低分辨质谱(ei-ms或者esi)以及高分辨质谱(hr-ms或者esi)使用finningan/mat-95仪器或者agilent6110质谱仪、orbitrap质谱仪记录。实验所用药品均为市售分析纯或化学纯,除特别说明所有试剂使用前均未经纯化处理。
实施例14-{6-[3-(4-氯-3-三氟甲基-苯基)-胍基]3-吡啶基-氧}-吡啶-2-羧酸甲胺(化合物1)的制备
化合物1
(1)4-(6-氨基吡啶-3-基)氧基)-n-甲基吡啶甲酰胺的制备
2-氨基-5-羟基吡啶5.00g(45.4mmol)溶解于n,n-二甲基乙酰胺中(40ml),加入叔丁醇钾6.11g(54.49mmol),于室温下搅拌30分钟,加入n-甲基-4-氯-2-吡啶甲酰胺7.75g(45.41mmol),于108摄氏度下反应18小时。将反应液冷却到室温,缓慢倒入冰水(100ml),用乙酸乙酯(3×40ml)萃取,有机层相用无水硫酸钠干燥,旋转蒸发蒸除溶剂,硅胶柱层析分离得4-(6-氨基吡啶-3-基)氧基)-n-甲基吡啶甲酰胺为白色固体9.80g,收率88%。
1hnmr(400mhz,d6-dmso)δh8.8(1h,d,j=5.1hz),8.5(1h,d,j=5.6hz),7.8(1h,dd,j=3.0,0.6hz),7.4(1h,d,j=2.6hz),7.3(1h,dd,j=8.9,3.0hz),7.1(1h,dd,j=5.6,2.6hz),6.6(1h,dd,j=8.9,0.7hz),6.1(2h,s),2.8(3h,d,j=4.8hz).ms(esi)[m+h]+:m/z245.1
(2)4-{6-[3-(4-氯-3-三氟甲基-苯基)-硫脲基]3-吡啶基-氧}-吡啶-2-羧酸甲胺的制备
将4-(6-氨基吡啶-3-基)氧基)-n-甲基吡啶甲酰胺5.00g(20.47mmol)溶于二氯甲烷(30ml),于室温下搅拌,缓慢加入4-氯-3-三氟甲基硫氰酸苯酯4.86g(20.27mmol),搅拌24小时。将反应液冷却到室温,缓慢倒入冰水(50ml),用二氯甲烷(3×30ml)萃取,有机层相用无水硫酸钠干燥,旋转蒸发蒸除溶剂,硅胶柱层析分离得4-{6-[3-(4-氯-3-三氟甲基-苯基)-硫脲基]3-吡啶基-氧}-吡啶-2-羧酸甲胺为白色固体9.00g,收率91%。
1hnmr(400mhz,chloroform-d)δh13.4(1h,s),9.1(1h,s),8.5(1h,d,j=5.5hz),8.2(1h,d,j=2.8hz),8.1–8.0(1h,m),8.0(1h,d,j=2.6hz),7.9(1h,dd,j=8.6,2.6hz),7.7(1h,d,j=2.6hz),7.6–7.5(2h,m),7.1(1h,dd,j=5.6,2.6hz),7.1–6.9 (1h,m),3.0(4h,d,j=5.1hz).ms(esi)[m+h]+:m/z482.1
(3)4-{6-[3-(4-氯-3-三氟甲基-苯基)-胍基]3-吡啶基-氧}-吡啶-2-羧酸甲胺(化合物1)的制备
将2-碘酰基苯甲酸2.09g(7.47mmol)溶于8ml饱和氨水溶液中,逐渐滴加到4-{6-[3-(4-氯-3-三氟甲基-苯基)-硫脲基]3-吡啶基-氧}-吡啶-2-羧酸甲胺2.00g的乙腈(15ml)溶液中,室温下搅拌3小时。将反应液倒入冰水(20ml),乙酸乙酯(3×20ml)萃取,有机层相用无水硫酸钠干燥,旋转蒸发蒸除溶剂,硅胶柱层析分离得白色固体1.50g,收率77%。
1hnmr(400mhz,chloroform-d)δh8.6(3h,s),8.4(1h,d,j=5.6hz),8.0(2h,dd,j=6.8,4.0hz),7.6(1h,d,j=2.4hz),7.5(1h,d,j=2.5hz),7.5–7.4(3h,m),7.2(1h,d,j=8.9hz),7.0(1h,dd,j=5.6,2.6hz),2.9(3h,d,j=5.0hz).ms(esi)[m+h]+:m/z465.1
实施例24-{4-[3-(4-氯-3-三氟甲基-苯基)-胍基]3,5-二氟苯氧基}-吡啶-2-羧酸甲胺(化合物2)的制备
化合物2
参照化合物1制备方法得到化合物2为白色固体41mg,收率78%。
1hnmr(400mhz,methanol-d4)δh8.5(1h,d,j=5.6hz),8.0(1h,d,j=2.6hz),7.6(2h,dt,j=5.8,2.9hz),7.5(1h,d,j=8.7hz),7.2(1h,dd,j=5.6,2.5hz),6.9(2h,d,j=7.7hz),3.0(3h,s).ms(esi)[m+h]+:m/z500.1
实施例34-{4-[3-(4-氯-3-三氟甲基-苯基)-胍基]3-氟苯氧基}-吡啶-2-羧酸甲胺(化合物3)的制备
化合物3
参照化合物1制备方法得化合物3为白色固体50mg,收率80%。
1hnmr(400mhz,chloroform-d)δh8.4(1h,d,j=5.5hz),8.1(1h,q,j=5.1hz),7.7–7.6(2h,m),7.5(1h,s),7.4(2h,s),7.1(1h,dd,j=5.6,2.5hz),6.9(2h,d,j=8.8hz),5.2(3h,d,j=129.8hz),3.0(3h,d,j=5.0hz).ms(esi)[m+h]+:m/z482.1
实施例44-{4-[3-(4-氯-3-三氟甲基-苯基)-胍基]苯氧基}-吡啶-2-羧酸甲胺(化合物4)的制备
化合物4
参照化合物1制备方法得到化合物4为白色固体41mg,收率78%。
1hnmr(400mhz,chloroform-d)δh8.4(1h,d,j=5.6hz),8.1(1h,q,j=5.1hz),7.6(1h,d,j=2.5hz),7.4(1h,d,j=2.6hz),7.4(1h,d,j=8.6hz),7.3(2h,d,j=6.0hz),7.2(1h,dd,j=8.6,2.5hz),7.1(1h,dd,j=5.6,2.6hz),7.0–7.0(2h,m),3.0(3h,d,j=5.1hz).ms(esi)[m+h]+:m/z464.2
实施例54-{4-[3-(2-(4-甲氧苯基-乙酰基)胍基]苯氧基}-n-甲基吡啶甲酰胺(化合物5)的制备
化合物5
(1)2-(4-甲氧基苯基)乙酰基异硫氰酸酯的制备
将对甲氧基苯乙酸2-(4-甲氧苯基)乙酸500mg(3.01mmol)溶于甲苯(10ml),加入二氯亚砜1.79g(15.04mmol),加热回流3小时后,旋转蒸发蒸除溶剂和二氯亚砜。将底物溶于无水四氢呋喃中,加入kscn520mg(5.3mmol),室温下搅拌12小时后旋转蒸发蒸除溶剂,得到棕色固体490mg,收率79%。因其不稳定,直接加入到下一步反应中。
(2)4-{4-[3-(2-(4-甲氧苯基-乙酰基)硫脲基]苯氧基}-n-甲基吡啶甲酰胺的制备
将2-(4-甲氧基苯基)乙酰基异硫氰酸酯100mg(0.56mmol)溶于无水二氯甲烷(5ml),加入4-(4-氨基苯氧基)-n-甲基-2-吡啶甲酰胺135mg(0.56mmol),于室温下搅拌3小时。旋转蒸发有机溶剂,硅胶柱层析分离得到4-{4-[3-(2-(4-甲氧苯基-乙酰基)硫脲基]苯氧基}-n-甲基吡啶甲酰胺白色固体190mg,收率75%。
1hnmrδh(400mhz,dmso-d6)δh12.4(1h,s),11.7(1h,s),8.8(1h,q,j=4.8hz),8.5(1h,d,j=5.6hz),7.7(2h,d,j=8.9hz),7.4(1h,d,j=2.6hz),7.3(4h,dd,j=8.6,6.1hz),7.2(1h,dd,j=5.6,2.6hz),6.9(2h,d,j=8.6hz),3.7(5h,s),2.8(3h,d,j=4.8hz).ms(esi)[m+h]+:m/z451.2
(3)4-{4-[3-(2-(4-甲氧苯基-乙酰基)胍基]苯氧基}-n-甲基吡啶甲酰胺(化合物5)的制备
4-{4-[3-(2-(4-甲氧苯基-乙酰基)硫脲基]苯氧基}-n-甲基吡啶甲酰胺80mg(0.17mmol)溶于乙腈(5ml),于室温下加入hmds286mg(1.78mmol),加入4-{4-[3-(2-(4-甲氧苯基-乙酰基)硫脲基]苯氧基}-n-甲基吡啶甲酰胺68mg于室温下搅拌。2小时后将反应液倒入冰水(15ml),乙酸乙酯(3×15ml)萃取,有机层相用无水硫酸钠干燥,旋转蒸发蒸除溶剂,硅胶柱层析分离得化合物5白色固体50mg,收率64%。
1hnmr(400mhz,dmso-d6)δh8.8(1h,q,j=4.8hz),8.5(1h,d,j=5.6hz),7.4(1h,d,j=2.6hz),7.2(2h,d,j=8.4hz),7.2(1h,dd,j=5.6,2.6hz),7.1(2h,d,j=8.8hz),6.9(1h,d,j=8.6hz),3.7(3h,s),3.5(2h,s),2.8(3h,d,j=4.8hz).ms(esi)[m+h]+:m/z434.2
实施例6n-甲基-4-(4-(3-特戊酰胍基)苯氧基)吡啶甲酰胺(化合物6)的制备
化合物6
参照化合物5制备方法得到化合物6为白色固体52mg,收率78%。
1hnmr(400mhz,chloroform-d)δh8.4(1h,d,j=5.6hz),8.1(1h,d,j=5.6hz),7.6(1h,d,j=2.5hz),7.2(2h,d,j=8.5hz),7.1(2h,d,j=8.8hz),7.0(1h,dd,j=5.6,2.5hz),3.0(3h,d,j=5.1hz),1.3(9h,s).ms(esi)[m+h]+:m/z370.2
实施例74-(4-(3-(3-氟苯甲酰基)胍基)苯氧基)-n-甲基吡啶甲酰胺(化合物7)的制备
化合物7
参照化合物5制备方法得到化合物7为白色固体42mg,收率72%。
1hnmr(400mhz,chloroform-d)δh8.4(1h,d,j=5.6hz),8.1(1h,d,j=5.2hz),7.9(1h,dt,j=7.8,1.2hz),7.8(1h,ddd,j=9.9,2.7,1.4hz),7.7(1h,d,j=2.5hz),7.4–7.3(3h,m),7.1(1h,s),7.1(2h,d,j=8.8hz),7.0(1h,dd,j=5.6,2.hz6),3.0(3h,d,j=5.1hz).ms(esi)[m+h]+:m/z408.2
实施例84-(4-(3-(4-氟苯甲酰基)胍基)苯氧基)-n-甲基吡啶甲酰胺(化合物8)的制备
化合物8
参照化合物5制备方法得到化合物8为白色固体41mg,收率71%。
4-(4-(3-(4-fluorobenzoyl)guanidino)phenoxy)-n-methylpicolinamide
1hnmr(400mhz,chloroform-d)δh8.4(1h,d,j=5.6hz),8.2(2h,dd,j=8.5,5.6hz),8.1(1h,d,j=5.9hz),7.7(1h,d,j=2.6hz),7.4(2h,d,j=8.2hz),7.2–7.0(5h,m),3.0(3h,d,j=5.1hz).ms(esi)[m+h]+:m/z408.2
实施例94-(4-(3-甲酰基胍基)苯氧基)-n-甲基吡啶甲酰胺(化合物9)的制备
化合物9
参照化合物5制备方法得到化合物9为白色固体58mg,收率81%。
1hnmr(400mhz,chloroform-d)δh8.4(1h,d,j=5.6),8.2–8.1(3h,m),7.7(1h,d,j=2.5),7.4(1h,d,j7.4),7.4–7.4(2h,m),7.3(1h,s),7.3(1h,s),7.1–7.0(2h,m),7.0(1h,dd,j=5.5,2.5),3.0(3h,d,j=4.9).ms(esi)[m+h]+:m/z390.2
实施例104-(4-(3-(4-氯-3-(三氟甲基)苯甲酰)胍基)苯氧基)-n-甲基吡啶甲酰胺(化 合物10)的制备
化合物10
参照化合物5制备方法得到化合物10为白色固体51mg,收率75%。
1hnmr(400mhz,dmso-d6)δh9.6(1h,s),8.8(1h,d,j=5.1hz),8.5(1h,d,j=5.6hz),8.5(1h,d,j=1.9hz),8.3(1h,dd,j=8.3,2.0hz),7.8(1h,d,j=8.3hz),7.6(2h,d,j=8.8hz),7.5(1h,d,j=2.6hz),7.2(2h,d,j=8.8hz),7.2(1h,dd,j=5.6,2.6hz),2.8(3h,d,j=4.8hz).ms(esi)[m+h]+:m/z492.1
化合物抗肿瘤活性测试实施例:
1、各化合物对骨髓瘤细胞(oci-my5、opm2、h929)和淋巴瘤细胞(molt-4)的杀伤活性:
1)实验方法:
不同浓度化合物处理细胞72h,cck-8法检测细胞存活率。
2)实验结果:
部分化合物的抗肿瘤活性
3)实验结论:
本发明的芳基胍化合物有一定的抗肿瘤活性,尤其是代表性化合物1与阳性药索拉菲尼(sorafenib)相当,进一步,我们测试了该化合物的动物活性。
2、化合物1的动物活性:
1)实验方法:
5~6周龄的雄性裸鼠6只,在右侧腋窝下种植h929细胞(2×106),大约两周后当肿瘤大小长到5mm×5mm时,随机将6只小鼠分成两组,每组3只,一组尾静脉注射化合物1(30mg/kg,2天1次),另一组尾静脉注射25%dmso的生理盐水,做为对照组。每两天测量肿瘤的大小,20天后,处死小鼠,取出肿瘤,称重、量大小。
2)实验结果:
20天后处死小鼠,取出肿瘤拍照,见图1。每两天测量肿瘤体积的大小,制作线形图,见图2。
3)实验结论:
本发明的芳基胍化合物在动物体内表现出一定的抗肿瘤活性,有进一步开发成抗肿瘤药物的潜力。
- 还没有人留言评论。精彩留言会获得点赞!