钝鳞紫背苔黄酮六位羟基氧甲基转移酶及其编码基因与应用的制作方法
本发明涉及基因工程技术领域,具体涉及一种钝鳞紫背苔黄酮六位羟基氧甲基转移酶及其编码基因与应用。
背景技术:
千层纸素a和高车前素是黄芩素和野黄芩素的6-oh甲基化的产物,具有广泛的生物活性,如抗氧化、抗真菌、抗炎、抗肿瘤等。有文献报道千层纸素a是目前临床上使用的抗分娩药物安宫宁中的主要有效活性成分。高车前素是传统药物灯盏花乙素在体内的活性代谢产物,但其具有更强的生物活性,口服易于吸收,生物利用度较灯盏花乙素提高了3倍多,具有更强的抗肿瘤、抗病毒、抗肝纤维化、抗老年痴呆、治疗缺血性脑血管病等作用。千层纸素a和高车前素有望成为新药的先导化合物,其合成也受到越来越广泛的关注。
但是,千层纸素a和高车前素在植物中含量不高,从植物中提取工艺流程复杂,分离纯化困难。有研究通过黄芩素和野黄芩素化学合成千层纸素a和高车前素,合成过程中为保护苯环上其余羟基不被甲基化,合成步骤复杂,合成时间长且需要多种辅助化合物的参与。
钝鳞紫背苔(plagiochasmaappendiculatum),是一种苔类植物,结构简单,没有茎叶分化,只有扁平的叶状体,没有真正的根和维管束。目前,对于钝鳞紫背苔的研究相对较少,从钝鳞紫背苔中分离鉴定出能够特异性催化黄酮六位羟基甲基化的氧甲基转移酶还未见报道。
技术实现要素:
针对上述现有技术,本发明的目的是提供钝鳞紫背苔黄酮六位羟基氧甲基转移酶及其编码基因与应用。该氧甲基转移酶来自于苔类植物钝鳞紫背苔,属于ⅰ类氧甲基转移酶(o-methyltransferases,omts),具有镁离子依赖性,是第一个能够高效催化黄酮六位羟基甲基化的氧甲基转移酶,可用于酶法合成千层纸素a和高车前素,其酶活反应条件简单温和,时间短,产物单一,转化率高。
为实现上述目的,本发明采用下述技术方案:
本发明提供的一种钝鳞紫背苔黄酮六位羟基氧甲基转移酶(paf6omt),其氨基酸序列为以下之一:
(1)seqidno.1所示的氨基酸序列;或
(2)将seqidno.1所示的氨基酸序列经过一个或几个氨基酸残基的取代和/或缺失和/或添加且功能相同的蛋白质。
上述钝鳞紫背苔黄酮六位羟基氧甲基转移酶的编码基因也属于本发明的保护范围。
上述编码基因(paf6omt)中,其核苷酸序列为以下之一:
(1)seqidno.2所示的核苷酸序列;或
(2)与seqidno.2所示的核苷酸序列具有90%以上同源性且编码相同功能蛋白质的核苷酸序列。
进一步的,本发明还提供了含有上述编码基因的表达载体。
上述表达载体为重组原核表达载体。
在本发明的一个具体实施方式中,所述重组原核表达载体为:在表达载体pet32a中插入上述编码基因。
本发明还提供了含有上述表达载体的重组细胞、转化体。
优选的,所述重组细胞为含有上述表达载体的细胞bl21(de3)。
本发明还提供用于扩增上述编码基因的引物对,其核苷酸序列分别如seqidno.3和seqidno.4所示。
上述钝鳞紫背苔黄酮六位羟基氧甲基转移酶在合成千层纸素a和/或高车前素中的应用也属于本发明的保护范围。
上述钝鳞紫背苔黄酮六位羟基氧甲基转移酶在催化黄芩素合成千层纸素a中的应用也属于本发明的保护范围。
上述钝鳞紫背苔黄酮六位羟基氧甲基转移酶在催化野黄芩素合成高车前素中的应用也属于本发明的保护范围。
本发明的有益效果:
本发明首次从钝鳞紫背苔转录组数据库中筛选出一个氧甲基转移酶,利用pcr成功扩增出其全长序列。通过构建pet32a蛋白表达载体转化大肠杆菌bl21(de3)得到目的蛋白。体外酶活功能鉴定证明paf6omt最适底物是黄芩素和野黄芩素。反应时间和蛋白量对底物转化率的影响的实验证明在合适的蛋白量和一定的反应时间内,蛋白可完全催化黄芩素和野黄芩素分别生成千层纸素a和高车前素。蛋白paf6omt可应用于酶法合成千层纸素a和高车前素,具有较高的应用价值和广阔的应用前景。
附图说明
图1:paf6omt基因全长cdna扩增产物的电泳图。
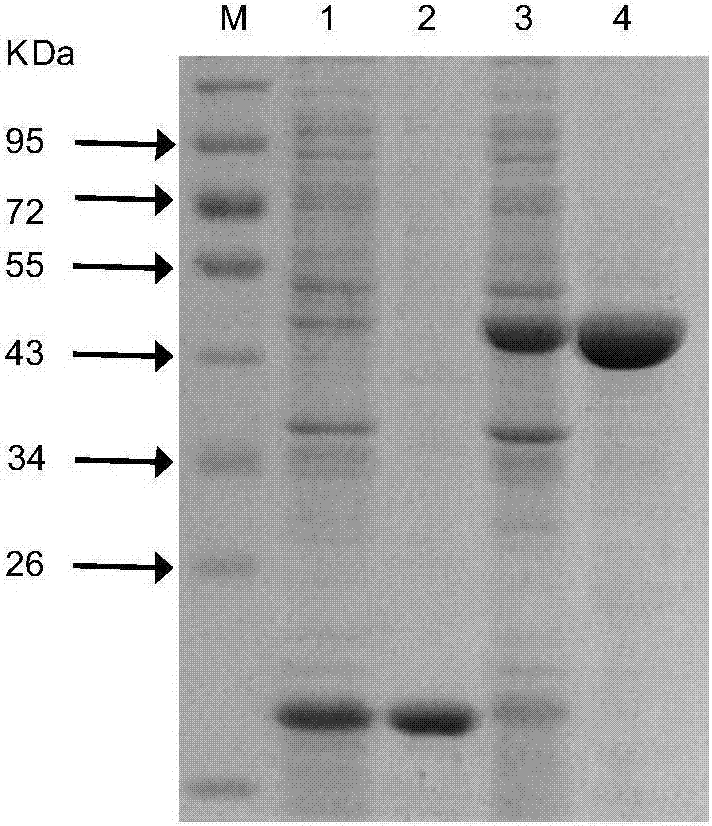
图2:paf6omt蛋白sds-page电泳图;
其中:m:蛋白分子质量标准
泳道1:空载体pet32a蛋白上清;
泳道2:空载体pet32a蛋白纯化;
泳道3:paf6omt蛋白上清;
泳道4:paf6omt蛋白纯化.
图3:hplc对反应产物的鉴定,反应底物为黄芩素和野黄芩素;
其中:a、d为空载体对照;
b、e为paf6omt蛋白酶活反应结果;
c、f为产物标准品。
图4:温度对酶活反应速率的影响。
图5a:蛋白量对底物转化率的影响;
图5b:反应时间对底物转化率的影响。
图6:paf6omt的亚细胞定位图;
其中:a:激发光下绿色荧光信号;b:激发光下叶绿体的荧光信号
c:自然光下烟草表皮细胞;d:a、b、c图的叠加。
具体实施方式
结合实施例对本发明作进一步的说明,应该说明的是,下述说明仅是为了解释本发明,并不对其内容进行限定。
下述实施例中,所使用的实验方法,未做具体说明的,均为常规方法,例如可以参考《分子克隆实验指南》(sambrook和russell,2001)。
下述实施例中,所使用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1.表达基因paf6omt的克隆
1.1ctab-pvp法提取钝鳞紫背苔总rna
(1)取新鲜的钝鳞紫背苔植物材料,清洗干净,用滤纸吸干多余水分,置于研钵中加液氮研磨至材料成粉末状。
(2)取适量粉末至提前预冷的离心管中,加800μl65℃预热的ctab-pvp提取液,上下颠倒混匀。
上述ctab-pvp提取缓冲液配制方法如下:
100mmtris·hcl(ph8.0),2%ctab(w/v),2%pvp(w/v),25mmedta,2mnacl,高压蒸汽灭菌之后巯基乙醇加至0.2%;溶液配置用depc处理过的ddh2o,高压灭菌后备用。
(3)65℃温浴30min,期间每隔6-10min颠倒混匀一次。
(4)冷却后加入600μl氯仿,混匀;4℃13,000rpm离心10min。
(5)取上清转至新离心管,加入600μl氯仿,振荡混合均匀后4℃13,000rpm离心10min。
(6)重复上步(即用氯仿抽提三次)。
(7)小心吸取上清转至新的1.5ml离心管中,加入1/3体积的8mlicl,-20℃静置3h以上(或4℃静置过夜)。
(8)4℃13,000rpm离心10min,弃上清。
(9)加入800μl75%乙醇洗涤沉淀2-3次。离心弃上清后挥干剩余乙醇。
(10)加入40μlproteinasek处理后的灭菌水溶解rna,制得总rna。使用biophotometerplus核酸蛋白测定仪测定提取的rna浓度及质量。
1.2paf6omt基因全长扩增
1.2.1引物设计
利用软件bioxm2.0的sequenceanalysis程序找出paf6omt的开放阅读框(openreadingframe)。在orf两侧的非编码区(untranslatedregion,utr)用软件primerpremier5.0设计全长引物paf6omt-f/r,对基因进行扩增;
全长引物:
paf6omt-f:gcattggaaggattgtttgg;(seqidno.3)
paf6omt-r:aattcgcttggcatcg;(seqidno.4)
1.2.2cdna合成
以提取的钝鳞紫背苔的总rna为模板,以primerscriptrtmastermix逆转录体系通过pcr技术获得cdna模板链。
逆转录体系及逆转录程序如下:
逆转录程序:37℃,15min;85℃变性15s,4℃保温。
逆转录产物保存于-20℃,使用前稀释10倍。
1.2.3目的基因扩增
以逆转录的钝鳞紫背苔cdna为模板,以paf6omt-f/r为引物进行扩增。
扩增体系及扩增程序如下:
pcr扩增程序:94℃预变性3min;94℃变性10s,50℃退火15s,72℃延伸30s,33个循环;72℃延伸10min。
pcr结果进行琼脂糖凝胶电泳检测(结果见图1),将目的大小条带切胶回收,方法如下。
将上述pcr产物进行琼脂糖凝胶电泳(1.5%,w/v,g/100ml),并用omega胶回收试剂盒回收目的片段。步骤如下:
(1)将上述pcr产物进行琼脂糖凝胶电泳(1.5%,w/v,g/100ml)后,经溴化乙锭(ethidiumbromide,eb)染色5min,于紫外灯下快速切下含有目的大小条带的胶块,放入1.5ml离心管。
(2)加入400μlbindingbuffer(xp2),在55℃水浴溶解胶块。期间每隔2-3min颠倒震荡离心管一次。
(3)将上述溶胶液转移至hibinddnacolumn,hibinddnacolumn置于2ml的收集管中,12,000rpm离心1min。
(4)弃掉收集管中的滤液,向hibinddnacolumn中加入300μlbindingbuffer(xp2)。12,000rpm离心1min,弃掉滤液。
(5)加入700μlspwwashbuffer,12,000rpm离心1min。
(6)重复上步一次。
(7)弃掉滤液,将空hibinddnacolumn室温12,000rpm离心2min挥干剩余乙醇。
(8)将hibinddnacolumn置于新的1.5ml离心管中,开盖放置至乙醇挥干。在柱子膜中央加入30μlddh2o,室温静置2min,12,000rpm离心1min。
(9)使用biophotometerplus核酸蛋白测定仪测定胶回收产物浓度及质量,回收片段立即使用或-20℃保存。
1.3目的片段的t-a克隆
1.3.1加a连t
将上述胶回收产物片段进行加a反应,体系及条件如下:
将各组分混匀,置于pcr仪中72℃反应40min后,将加a产物与t载体(购自takara公司)进行连接,反应体系如下:
轻轻吹打混合内容物,短暂离心,16℃连接3h。
1.3.2转化
将上述10μl连接产物转化大肠杆菌dh5α感受态。转化方法如下:-80℃保存的大肠杆菌dh5α感受态细胞(50μl)取出后置于冰上解冻,加入10μl连接产物,轻轻吹打混匀,冰上静置30min;42℃水浴中热击90s后迅速置于冰上2min,加入600μllb培养基后于37℃培养箱振荡培养1h,取200μl转化液涂于lb固体培养基(含100μg/mlamp)上,37℃静置培养12h。
lb培养基组分(1l):酵母提取物5g、胰蛋白胨10g、nacl10g,加水溶解后,调ph至7.0,定容。固体培养基加入琼脂(12g/l)后,高压蒸汽灭菌。
1.3.3重组子阳性克隆鉴定
37℃培养12h后,随机挑选12个单克隆于200μllb培养基,37℃振荡培养4h。以m13f/r为引物,菌液为模板进行菌落pcr。体系如下:
扩增程序:94℃预变性5min;94℃变性30s,56℃退火30s,72℃延伸60s,32个循环;72℃延伸10min;
菌落pcr后进行琼脂糖凝胶电泳,将大小合适的阳性克隆送博尚生物技术有限公司测序。阳性克隆存菌:菌液930μl加dmso70μl,混和均匀后于-80℃保存。
实施例2.基因蛋白表达及酶活功能分析
2.1目的基因原核表达载体的构建
根据目的基因paf6omt及pet32a载体的序列信息,选择合适的酶切位点设计原核表达载体的引物paf6omt-pet32a-f与paf6omt-pet32a-r:
paf6omt-pet32a-f:cgggatccatggccccagaggttttgtc;(seqidno.5)
paf6omt-pet32a-r:ccctcgagctacttaagagttgaaatga;(seqidno.6)
以pmd19t-paf6omt质粒为模板,用上述引物扩增其orf,pcr产物电泳并胶回收,载体pet32a及胶回收片段用bamhi与xhoi进行酶切,酶切体系如下:
酶切在37℃水浴进行,反应3h。酶切产物加10×loadingbuffer终止反应后进行琼脂糖凝胶电泳并选择合适的条带进行胶回收,胶回收方法同上。
2.1.1连接
酶切后的目的片段与酶切后的载体pet32a(购自novagen公司)用t4dnaligase(购自takara公司)连接,体系如下:
将上述组分充分混匀后,16℃过夜连接。
2.1.2转化
上述连接产物转化大肠杆菌dh5α感受态。转化方法同上。
2.1.3重组子阳性克隆鉴定
菌落pcr验证阳性,以paf6omt-pet32a-f/r为引物,验证方法同上。
选出目的大小的阳性克隆送博尚生物技术有限公司测序,同时扩大培养抽提质粒,进行酶切验证。若能切下与目的片段大小相符的条带则证明该克隆为阳性。至此,载体构建完成。
构建完成后摇菌提paf6omt-pet32a的质粒,备用。
2.1.4转化
构建好的原核表达载体质粒热击法转化大肠杆菌bl21(de3)感受态细胞,转化、筛选与鉴定方法同上。
2.2基因重组蛋白原核表达
2.2.1重组蛋白诱导纯化
(1)挑取paf6omt-pet32a-bl21阳性克隆接种于4ml培养基(含amp抗性)中,37℃过夜震荡培养。
(2)菌液按1:100接种于100ml培养基(含amp抗性)中,37℃震荡培养至od6000.4-0.6。加100μl1m的iptg,在18℃,110rpm摇床里培养18h诱导蛋白表达。
(3)50ml离心管收集菌液,5000rpm,5min离心后弃上清。
(4)bindingbuffer重悬菌体,5000rpm,5min离心后弃上清。洗涤两次后加20mlbindingbuffer重悬菌体。
(5)超声裂解菌体,4℃12,000rpm,离心20min后收集上清进行过柱纯化。
(6)先加10mlbindingbuffer洗脱杂蛋白,再加4mlbindingbuffer和elutionbuffer的1:1混合溶液进行洗脱,收集洗脱液。洗脱液置于超滤管中进行换液并浓缩。
(7)将浓缩液吸取至10ml收集管中,测定蛋白浓度,并留样电泳。
(8)蛋白加20%甘油后,保存至-80℃冰箱备用。
bindingbuffer:分别称取2.42gtris-hcl、29.22gnacl、0.34gimidazole,加水溶解,定1000ml,灭菌后加入70μlβ-巯基乙醇,4℃保存。
elutionbuffer:分别称取2.42gtris-hcl、29.22gnacl、34gimidazole,加水溶解,定容 1000ml,灭菌后加入70μlβ-巯基乙醇,4℃保存。
2.2.2蛋白sds-page电泳
目的蛋白的表达及分离纯化情况用变性的聚丙烯酰胺凝胶电泳(sodiumdodecylsulfatepolyacrylamidegelelectrophoresis,sds-page)进行检测。
(1)组装电泳装置,将玻璃板固定于胶架上。
(2)配制12%分离胶,加入到电泳仪中,加水液封,静置直至分离胶凝固。
(3)配制5%浓缩胶,倒掉上层水。将配制好的5%的浓缩胶混匀后立即倒入,将孔梳插入到玻璃板间(避免气泡的产生),待胶凝固后拔出梳子。
(4)在蛋白上清液和纯化的蛋白中分别加入适量loadingbuffer,在沸水中煮5min13,000rpm离心10min,取上清20μl点样,同时吸取蛋白marker3μl点样。
(5)向电泳槽中加适量电泳缓冲液,先90v恒压进行电泳,待样品电泳到分离胶时改为160v恒压电泳,直至溴酚蓝至胶的下沿,停止电泳。
(6)取下蛋白胶,放入考马斯亮蓝r-250染色液中浸泡染色,室温下轻摇染色2h。
(7)用蒸馏水冲洗掉蛋白胶表面的染色液,冲洗2~3次后置于脱色液中脱色2h,脱色过程中更换脱色液数次,直至蛋白胶背景洗干净。
(8)观察结果,照相保存并进行分析,蛋白电泳结果如图2。
2.3蛋白体外酶活功能鉴定
2.3.1蛋白最适底物筛选
paf6omt进行体外酶活功能鉴定,筛选蛋白最适底物。底物有野黄芩素、黄芩素、槲皮素、木犀草素、圣草酚、秦皮乙素、咖啡酰辅酶a、咖啡醇、咖啡醛、咖啡酸、5-oh阿魏酸、5-oh松柏醇、儿茶酚。酶活反应体系如下:
37℃水浴,酶活反应30min后加等体积乙腈终止反应。以pet32a空载体蛋白的酶活反应作对照,采用hplc进行酶活反应分析。
2.3.2酶活产物分析
为了验证paf6omt的体外酶活功能,hplc被用于检测上述酶活反应的产物。分析采用eclipsexd-c18,5μm,4.6×150mm(agilent)色谱柱,检测波长254nm,280nm,320nm和346nm,流速1.0ml/min。液相分析条件如下:
液相分析条件1
液相分析条件2
液相分析条件3
液相分析条件1用于检测野黄芩素、黄芩素,鉴定结果如图3;液相分析条件2用于检测槲皮素、木犀草素、圣草酚;液相分析条件3用于检测秦皮乙素、咖啡酰辅酶a、咖啡醇、咖啡醛、咖啡酸、5-oh阿魏酸、5-oh松柏醇、儿茶酚。酶活反应分析结果如表1。
表1:
aactivitiespresentedarenmol·(mg·min)-1±stdev.
bnoproductdetected.
2.3.3最适温度测定
选取黄芩素做底物,测定温度对paf6omt蛋白体外酶活的影响。最适温度的测定在ph7.5的tris-hcl缓冲液中进行,反应时间30min,温度梯度设定为25℃、30℃、35℃、37℃、40℃、45℃、50℃。测定结果如图4。
2.3.4酶学动力学参数测定
在tris-hcl(ph7.5)缓冲液中,37℃条件下,进行paf6omt的酶学动力学分析,将底物浓度分别设为10、20、40、50、80、100、150、200μm。反应在酶加入时开始计时,反应5min,加等体积乙腈终止反应,平行实验3次。实验结果见表2。
表2:
实施例3.paf6omt酶法合成高车前素和千层纸素aa
在体外酶活功能鉴定实验中,paf6omt蛋白对黄芩素和野黄芩素的6-oh的甲基化选择性很高,且反应产物单一,底物转化率高,可以用来酶法合成千层纸素aa和高车前素。
3.1蛋白量对底物转化率的影响
为了研究蛋白量对底物转化率的影响,我们在tris-hcl(ph7.5)缓冲液中,37℃条件下,设定蛋白量梯度1-14μg,反应时间1h。酶活体系同上,其中sam和mgci2的加入量随蛋白量的增加成比例增加。蛋白量对底物转化率的影响结果如图5a。
3.2反应时间对底物转化率的影响
在tris-hcl(ph7.5)缓冲液中,37℃条件下,固定蛋白量6μg,设定不同的时间梯度5min、10min、20min、30min、45min、60min、90min、2h、2.5h、3h、4h进行反应,用以检测反应时间对底物转化率的影响,结果见图5b。
实施例4.基因亚细胞定位
4.1目的基因gfp定位载体的构建
根据目的基因paf6omt设计引物paf6omt-gfpf与paf6omt-gfpr:
paf6omt-gfpf:cgggatccatggccccagaggttttgtc;(seqidno.7)
paf6omt-gfpr:ggggtacccttaagagttgaaatgacaa;(seqidno.8)
以paf6omt-pet32a质粒为模板,扩增其orf(去除终止子),pcr产物电泳并回收,载体p35s-1301-gfp及回收片段用bamhi与kpni进行酶切,酶切体系如下:
酶切在37℃水浴进行,反应3h。酶切产物进行琼脂糖凝胶电泳并选择合适的条带进行胶回收,胶回收方法同上。
4.1.1酶切产物的连接,方法同上
4.1.2连接转化及阳性克隆鉴定,方法同上
构建完成后摇菌提其质粒paf6omt-p35s-1301-gfp,转化农杆菌备用。
4.2冻融法转化农杆菌
(1)取出农杆菌感受态细胞eha105,冰上融化,分别加入2μl质粒 paf6omt-p35s-1301-gfp和空载体p35s-1301-gfp质粒,用移液枪轻轻吹打混匀,冰上放置25min;
(2)液氮速冻60s后,置于37℃水浴3min;
(3)加入400μl无抗性yep液体培养基,30℃振荡培养4h;
(4)取200μl菌液涂于yep固体培养基(含50mg/mlkan,100mg/mlrif)上。30℃静置培养36h;
(5)挑取单克隆进行小量振荡培养,菌落pcr鉴定其阳性,取阳性克隆存菌,备用。
yep培养基组分(1l):酵母提取物10g、胰蛋白胨10g、nacl5g,加水溶解并定容。固体培养基加入琼脂(12g/l)后,高压蒸汽灭菌。
4.3农杆菌瞬时转化法转化烟草表皮细胞
(1)将paf6omt-p35s-1301-gfp-eha105、p35s-1301-gf-eha105p和抑制蛋白沉默的p19划板,30℃培养36h后挑单克隆接种至2mlyep液体培养液中(含50mg/mlkan,100mg/mlrif)小摇,30℃振荡培养12h。
(2)将菌液按1:100接种于10mlyep液体培养液(含50mg/mlkan,100mg/mlrif)中大摇至od600在0.4-0.6。
(3)收菌,4000rpm,离心20min,弃上清;烟草转化液洗涤1次,离心,弃上清。
(4)菌体用少量转化液重悬,调od600≈1.0。
(5)将paf6omt-p35s-1301-gfp-eha105、p35s-1301-gfp-eha105分别和p19按1:1混合后置于25℃,110rpm,活化2h。
(6)用1ml的注射器将农杆菌渗透到烟草叶片的下表皮细胞中。
(7)36h后,取经农杆菌渗透的叶片在激光共聚焦显微镜上检测下表皮细胞的荧光信号(设置488nm的氩激发光,gfp信号的发射光波长495-570nm,叶绿体发出的信号波长650-760nm),结果见图6。
烟草转化液:mes-koh(ph5.6)、na3po42mm、葡萄糖0.5%(v/v)、乙酰丁香酮100μl。
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围以内。
- 还没有人留言评论。精彩留言会获得点赞!