四环化合物的制作方法
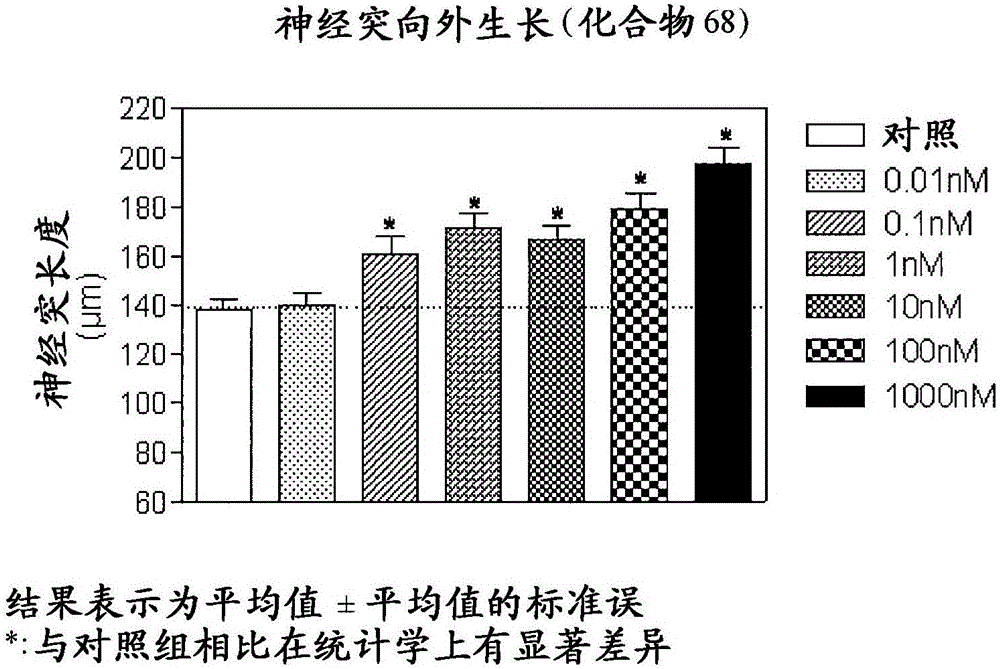
相关申请的交叉参考该申请要求2007年10月25日提交的U.S.临时专利申请60/982,678、2007年10月25日提交的U.S.临时专利申请60/982,679、2008年1月25日提交的U.S.临时专利申请61/062,332、2008年1月25日提交的U.S.临时专利申请61/062,331、2008年3月4日提交的U.S.临时专利申请61/033,754、2008年3月4日提交的U.S.临时专利申请61/033,757和2007年10月25日提交的俄罗斯专利申请2007139634的优先权,其各自的公开内容被全部引入本文作为参考。受联邦政府资助的研究项目中所做出的发明的权利的声明不适用。发明背景神经递质诸如组胺、血清素、多巴胺和去甲肾上腺素介导在中枢神经系统(CNS)之中以及CNS之外的大量进程。异常的神经递质水平与多种疾病和病症相关,其包括但不限于阿尔茨海默病、帕金森病、孤独症、吉兰巴雷综合征、轻度认知功能损害(mildcognitiveimpairment)、精神分裂症、焦虑、多发性硬化、中风、创伤性脑损伤、脊髓损伤、糖尿病性神经病、纤维肌痛、双相性精神障碍、精神病、抑郁症和多种变应性疾病。调节这些神经递质的化合物可以是有用的治疗剂。组胺受体属于G蛋白偶联7次跨膜跨膜蛋白的超家族。G蛋白偶联受体构成真核细胞中主要的信号转导系统之一。在被认为参与激动剂-拮抗剂结合位点的那些区域中的这些受体的编码序列在哺乳动物物种中是高度保守的。在大多数外周组织中及中枢神经系统中发现了组胺受体。能调节组胺受体的化合物可用于治疗中,例如组胺拮抗剂可用作抗阻胺药。Dimebon是一种已知的抗-组胺药,还被鉴定为一种神经保护剂,可用于治疗尤其是神经变性疾病。在阿尔茨海默病和亨廷顿病的临床前模型中Dimebon已显示出抑制脑细胞(神经元)的死亡,这使得它成为这些和其它的神经变性疾病的新的潜在的治疗方法。此外,在细胞应激情况下Dimebon显示可高效地改善细胞的线粒体功能。例如,用细胞毒素离子霉素处理后Dimebon治疗剂量依赖性地改善线粒体功能并增加存活细胞的数量。Dimebon还显示出促进神经突向外生长和神经发生、突起(其在形成新的和/或增强的神经元细胞连接中是重要的),以及Dimebon在用于另外的疾病或病症的潜能的证据。参见例如,U.S.专利6,187,785和7,071,206和PCT专利申请PCT/US2004/041081、PCT/US2007/020483、PCT/US2006/039077、PCT/US2008/077090、PCT/US2007/020516、PCT/US2007/022645、PCT/US2007/002117、PCT/US2008/006667、PCT/US2007/024626、PCT/US2008/009357、PCT/US2007/024623和PCT/US2008/008121。本文所公开的所有的参考文献诸如出版物、专利、专利申请和公布的专利申请的全部内容被引入本文作为参考。尽管Dimebon很有希望作为药物用于治疗神经变性疾病和/或其中在治疗中可能牵涉神经突向外生长和/或神经发生的疾病,但人们仍然需要用于治疗此类疾病或病症的新的和作为选择的治疗剂。此外,人们仍然需要新的和作为选择的抗组胺药,优选没有副作用(诸如嗜睡)或副作用减少的抗组胺药。相比于Dimebon显示出增强的和/或更期望性质(例如,更好的安全和有效性)的化合物可特别用于治疗至少那些认为Dimebon对其有益的适应症。而且,相比于Dimebon,通过例如体外和/或体内试验测定显示出不同治疗特性的化合物可用于另外的疾病和病症。发明概述已经合成并在生物化学和基于细胞的试验以及体内研究中测试了众多化合物。通式(I)的四环化合物被描述为新的组胺受体调节剂。其他的化合物在本文也有详述。本发明提供了包含该化合物的组合物,还提供了包含该化合物以及使用和制备该化合物的方法的药盒。还提供了其它的四环化合物。还发现本发明的化合物在治疗神经变性疾病中的用途。还发现本发明的化合物在治疗其中在治疗中可能牵涉调节胺能G蛋白偶联受体和/或神经突向外生长的疾病和/或病症中的用途。本文所公开的化合物可用于本文所公开的方法中,其包括在有需要的个体诸如人中,治疗、预防认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症,延缓其发作和/或延缓其发展的用途。在一个实施方案中,提供了式(I)化合物:其中:R1是H、羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基、羟基、烷氧基、硝基或R2a和R2b一起形成羰基部分;R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基或硝基或R3a和R3b一起形成羰基部分;X7、X8、X9和X10各自独立地是N或CR4;m和q独立地是0或1;X1是N或CH;R4各自独立地是H、羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基;R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H、羟基、C1-C8烷基或与其所连接的碳及一个孪位的R8(a-f)一起形成环烷基部分或羰基部分;R10a和R10b各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、羟基、烷氧基或R10a和R10b一起形成羰基部分;Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;条件是:(i)当X1是N,所述化合物不是表1中的化合物或其盐;(ii)所述化合物不是化合物229H、230、241、255、256、262和274中任何一种或其盐;且(iii)所述化合物不是在U.S.专利6,187,785、7,071,206、3,409,628和6,849,640或在PCT公开WO2005/055951、WO2007/041697和WO2007/087425中所列出的那些化合物。在另一个实施方案中,本发明化合物、其药物组合物、其被分离的形式及本文详述的使用所述化合物及施用所述化合物的方法包括式(I)化合物中的任何一个,其包括在表1所列的那些或其盐及化合物229H、230、241、255、256、262和274或其盐。在一个实施方案中,化合物是其中R4不是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、芳氧基和芳烷基的式(I)化合物。在一个实施方案中,化合物是其中R4不是被取代的或未被取代的芳基的式(I)化合物。在又一个实施方案中,化合物是其中X7-X9中至少一个是C-R4且R4不是H的式(I)化合物。在一个实施方案中,PCT公开WO2008/123800、WO2008/123796、WO2008/115098和WO2008/060190中的任何一个所列出的化合物从任何本文详述的化合物结构式中被排除,且将这些公开物的全部内容引入本文作为参考且尤其针对的是其中所详述的化合物。在一个实施方案中,PCT公开WO2008/123800,WO2008/123796、WO2008/115098和WO2008/060190中的任何一个所列出的化合物包括在任何本文详述的化合物结构式之中,且可用于本文提供的方法。在另一个实施方案中,化合物是式(I)化合物,其中进一步的条件是当Q是被取代的或未被取代的杂芳基时其不是噻唑、三唑或二唑。然而,在另一个实施方案中,化合物是式(I)化合物,且包括其中Q是噻唑、三唑或二唑的化合物。在另一个实施方案中,化合物是式(I)化合物,其中进一步的条件是所述化合物不是式(G)化合物。然而,在另一个实施方案中,化合物是式(I)化合物,且包括与式(G)一致的化合物。表1.表1A.在一个实施方案中,化合物是式(I)化合物,其中所述化合物进一步是1型化合物。在另一个实施方案中,化合物是式(I)化合物,其中所述化合物进一步是2型化合物。在另一个实施方案中,化合物是式(I)化合物,其中所述化合物进一步是3型化合物。在又一个实施方案中,化合物是式(I)化合物,其中所述化合物进一步是4型化合物。本发明还包括本文所涉及化合物的所有的盐,诸如可药用盐。本发明还包括所述化合物的任何或所有的立体化学形式(其包括任何的对映异构或非对映异构形式)和任何的互变异构体或其他形式。除非结构或名称中明确表明立体化学,否则结构或名称旨在包括所述化合物所有可能的立体异构体。此外,当描述一种特定的立体化学形式时,应理解本发明还包括其他的立体化学形式。本发明还包括化合物的所有形式,诸如化合物的结晶或非结晶形式。本发明还涉及包含本发明化合物的组合物,诸如基本上纯的化合物的组合物,所述化合物包括其特定的立体化学形式。包含以任何比例的本发明化合物的混合物的组合物也包括在本发明中,其包括以任何比例的本发明化合物的两种或多种立体化学形式的混合物,由此化合物的外消旋混合物、非外消旋混合物、对映体富集(enantioenriched)混合物和呈比例(scalemic)混合物包括在内。在一方面,本发明化合物用于在有需要的个体诸如人中治疗、预防以下各项中的任何一项或多项,延缓其发作和/或延缓其发展:认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症。在一个实施方案中,本发明化合物用于治疗、预防疾病或病症、延缓其发作和/或延缓其发展,其中认为调节胺能G蛋白偶联受体对所述疾病或病症有益或调节胺能G蛋白偶联受体对所述疾病或病症是有益的。在一个实施方案中,本发明化合物用于治疗、预防任何一种或多种疾病或病症、延缓其发作和/或延缓其发展,其中认为神经突向外生长和/或神经发生和/或神经营养作用对所述疾病或病症有益或神经突向外生长和/或神经发生和/或神经营养作用对所述疾病或病症是有益的。在另一个实施方案中,本发明化合物用于治疗、预防疾病或病症、延缓其发作和/或延缓其发展,其中认为调节胺能G蛋白偶联受体和神经突向外生长和/或神经发生和/或神经营养作用对对所述疾病或病症有益或调节胺能G蛋白偶联受体和神经突向外生长和/或神经发生和/或神经营养作用对所述疾病或病症是有益的。在一个实施方案中,所述疾病或病症是认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症。在另一个方面,本发明化合物用于在个体中改善认知功能和/或降低精神病效应,其包括向有需要的个体施用对改善认知功能和/或降低精神病效应有效的量的本文所述的化合物或其可药用盐。在另一个方面,本发明化合物用于在个体中刺激神经突向外生长和/或促进神经发生和/或增强神经营养作用,其包括向有需要的个体施用对刺激神经突向外生长和/或促进神经发生和/或增强神经营养作用有效的量的本文所述的化合物或其可药用盐。突触损失与多种神经变性疾病和病症相关,所述疾病和病症包括阿尔茨海默病、亨廷顿病、中风、头损伤和脊髓损伤。刺激神经突向外生长的本发明的化合物可对这些情况有益。在另一个方面,本文所述的化合物用于调节胺能G蛋白偶联受体,其包括向有需要的个体施用对调节胺能G蛋白偶联受体有效的量的本文所述的化合物或其可药用盐。在一个实施方案中,本发明化合物调节以下受体中的至少一种:肾上腺素能受体(例如,a1D、a2A和/或a2B)、血清素受体(例如,5-HT2A、5-HT2C、5-HT6和/或5-HT7)、多巴胺受体(例如,D2L)和组胺受体(例如,H1、H2和/或H3)。在另一个实施方案中,调节以下受体中的至少两种:肾上腺素能受体(例如,a1D、a2A和/或a2B)、血清素受体(例如,5-HT2A、5-HT2C、5-HT6和/或5-HT7)、多巴胺受体(例如,D2L)和组胺受体(例如,H1、H2和/或H3)。在另一个实施方案中,调节以下受体中的至少三种:肾上腺素能受体(例如,a1D、a2A和/或a2B)、血清素受体(例如,5-HT2A、5-HT2C、5-HT6和/或5-HT7)、多巴胺受体(例如,D2L)和组胺受体(例如,H1、H2和/或H3)。在另一个实施方案中,调节以下受体中的每一种:肾上腺素能受体(例如,a1D、a2A和/或a2B)、血清素受体(例如,5-HT2A、5-HT2C、5-HT6和/或5-HT7)、多巴胺受体(例如,D2L)和组胺受体(例如,H1、H2和/或H3)。在另一个实施方案中,调节以下受体中的至少一种:a1D、a2A、a2B、5-HT2A、5-HT2C、5-HT6、5-HT7、D2L、H1、H2和H3。在另一个实施方案中,调节以下受体中的至少二或三或四或五或六或七或八或九或十或十一种:a1D、a2A、a2B、5-HT2A、5-HT2C、5-HT6、5-HT7、D2L、H1、H2和H3。在一个特定的实施方案中,至少多巴胺受体D2L被调节。在另一个特定的实施方案中,至少多巴胺受体D2L和血清素受体5-HT2A被调节。在又一个特定的实施方案中,至少肾上腺素能受体a1D、a2A、a2B和血清素受体5-HT6被调节。在另一个特定的实施方案中,至少肾上腺素能受体a1D、a2A、a2B、血清素受体5-HT6和一种或多种血清素受体5-HT7、5-HT2A、5-HT2C和组胺受体H1和H2被调节。在又一个特定的实施方案中,调节组胺受体H1。在另一个实施方案中,本发明化合物展现本文详述的任何受体调节活性,并且还刺激神经突向外生长和/或神经发生和/或增强神经营养作用。本发明还涉及包含本发明化合物及可药用载体或赋形剂的药物组合物。包含本发明化合物及使用说明书的药盒也包括在本发明内。附图简述图1:图1A-D显示在使用皮质神经元的情况下不同浓度的化合物395、68、250和20对神经突向外生长的作用。星号(*)表示相对于对照组有显著性差异。图1E显示不同浓度的化合物395对在使用混合的皮质培养物的情况下神经突向外生长的作用。图2:图2A和2B显示了用不同浓度的化合物225治疗的东莨菪碱处理的大鼠中的识别得分和优秀学习者的百分比。图2C和2D显示了用不同浓度的化合物68治疗的东莨菪碱处理的大鼠中的识别得分和优秀学习者的百分比。图2E和2F用不同浓度的化合物395治疗的东莨菪碱处理的大鼠中的识别得分和优秀学习者的百分比。图3:图3A显示了在OF中在90分钟的测试期间氯氮平和化合物36对总行进路程的作用的时程。图3B和3C显示了氯氮平和化合物36对总行进路程的作用。数据是在PCP注射前(基线)30分钟内(B)的和,以及在PCP注射后60分钟内(C)的和,并且表示平均值±SEM。星号(*p<0.05)表示与DMSO-PCP相比的显著性差异。井号(#p<0.05)表示与溶媒-PCP相比的显著性差异。图3D显示了在90分钟的测试期间氯氮平和化合物36对总的站立(rearing)的作用的时程。数据表示平均值±SEM。图3E和3F显示了氯氮平和化合物36对总的站立的作用。数据是在PCP注射前(基线)30分钟内(E)的和,以及在PCP注射后60分钟内(F)的和,并且表示平均值±SEM。星号(*p<0.05)表示与DMSO-PCP相比的显著性差异。井号(#p<0.05)表示与溶媒-PCP相比的显著性差异。图4:图4A显示了在90分钟的测试期间氯氮平和化合物247对总行进路程的作用的时程。图4B和4C显示了氯氮平和化合物247对总行进路程的作用。数据是在PCP注射前(基线)30分钟内(B)的和,以及在PCP注射后60分钟内(C)的和,并且表示平均值±SEM。星号(*)表示与PBS相比的显著性差异。井号(#)表示与DMSO相比的显著性差异。图5A显示了对化合物C4-1、C4-5、C4-4而言的谷氨酸诱导的Ca摄入到突触小体内。图5B显示了对化合物C4-6、C4-7而言的谷氨酸诱导的Ca摄入到突触小体内。图6A显示了对化合物C1-1、C1-5而言的谷氨酸诱导的Ca摄入到突触小体内。图6B显示了对化合物C1-7而言的谷氨酸诱导的Ca摄入到突触小体内。图6C显示了对化合物C1-6而言的谷氨酸诱导的Ca摄入到突触小体内。图6D显示了对化合物C1-4、C1-8而言的谷氨酸诱导的Ca摄入到突触小体内。图7显示了对化合物C4-3、C2-1而言的谷氨酸诱导的Ca摄入到突触小体内。图8显示了C4-1、C4-5和C4-4对大鼠浦肯野细胞AMPA受体电流的作用。图9显示了C4-6和C4-7对大鼠浦肯野细胞AMPA受体电流的作用。图10显示了C1-1和C1-5对大鼠浦肯野细胞AMPA受体电流的作用。图11显示了Dimebon和环噻嗪对AMPA-受体电流的相比较的作用。图12显示了化合物C1-4、C1-6、C1-8和C4-3对红藻氨酸诱导的浦肯野细胞中的电流的作用。图13显示了化合物对Ca2+-诱导的线粒体混悬液的A540的降低的初始速率(Vi)的影响。图14显示了不同剂量的Dimebon对探究熟悉的和新的物体的持续时间的作用。图15显示了不同剂量的Dimebon对探究在熟悉的和新的位置的物体的持续时间的作用。图16显示了不同剂量的化合物C4-1对探究在熟悉的和新的位置的物体的持续时间的作用。图17显示了不同剂量的化合物C4-4对探究在熟悉的和新的位置的物体的持续时间的作用。图18显示了不同剂量的化合物C4-5对探究在熟悉的和新的位置的物体的持续时间的作用。图19显示了不同剂量的化合物C4-6对探究在熟悉的和新的位置的物体的持续时间的作用。图20显示了不同剂量的化合物C4-7对探究在熟悉的和新的位置的物体的持续时间的作用。图21显示了不同剂量的化合物C1-1对探究在熟悉的和新的位置的物体的持续时间的作用。图22显示了不同剂量的化合物C1-5对探究在熟悉的和新的位置的物体的持续时间的作用。发明详述定义除非另外清楚地说明,术语“一个”、“一种”等是指一个/种或多个/种。本文使用的术语“约”指本领域技术人员容易知晓的相应值的通常的变化范围。本文中提到“约”某一数值或参数时包括(并描述了)涉及该数值或参数本身的实施方案。本文使用的术语“胺能G蛋白偶联受体”是指参与细胞通信的跨膜蛋白家族。胺能G蛋白偶联受体通过生物胺类活化,并代表G蛋白偶联受体超家族的一个亚类,其结构通过7个跨膜螺旋来表征。胺能G蛋白偶联受体包括但不限于肾上腺素能受体、血清素受体、多巴胺受体、组胺受体和咪唑啉受体。本文使用的术语“肾上腺素能受体调节剂”是指且包括结合于或抑制配体结合于肾上腺素能受体或者降低或消除或增加或增强或模拟肾上腺素能受体活性的化合物。因此,“肾上腺素能受体调节剂”包括肾上腺素能受体拮抗剂和肾上腺素能受体激动剂。在一些方面,肾上腺素能受体调节剂以可逆的或不可逆的方式结合于或抑制配体结合于α1-肾上腺素能受体(例如,α1A、α1B和/或α1D)和/或α2-肾上腺素能受体(例如,α2A、α2B和/或α2C)和/或降低或消除或增加或增强或模拟α1-肾上腺素能受体(例如,α1A、α1B和/或α1D)和/或α2-肾上腺素能受体(例如,α2A、α2B和/或α2C)的活性。在一些方面,肾上腺素能受体调节剂抑制配体结合的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%,如在本文所述的试验中所测定的那样。在一些方面,与在相同个体中用肾上腺素能受体调节剂治疗之前的相应的活性相比,或与在没有接受肾上腺素能受体调节剂的其他个体中的相应的活性相比,肾上腺素能受体调节剂降低肾上腺素能受体的活性的至少或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%。在一些方面,与在相同个体中用肾上腺素能受体调节剂治疗之前的相应的活性相比,或与在没有接受肾上腺素能受体调节剂的其他个体中的相应的活性相比,肾上腺素能受体调节剂增强肾上腺素能受体的活性的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%或200%或300%或400%或500%或更多。在一些方面,肾上腺素能受体调节剂能够结合于肾上腺素能受体的活性位点(例如,配体的结合位点)。在一些实施方案中,肾上腺素能受体调节剂能够结合于肾上腺素能受体的别构位点。本文使用的术语“多巴胺受体调节剂”是指且包括结合于或或抑制配体结合于多巴胺受体或者降低或消除或增加或增强或模拟多巴胺受体活性的化合物。因此,“多巴胺受体调节剂”包括多巴胺受体拮抗剂和多巴胺受体激动剂。在一些方面,多巴胺受体调节剂以可逆的或不可逆的方式结合于或抑制配体结合于多巴胺-1(D1)和/或多巴胺-2(D2)受体或者降低或消除或增加或增强或模拟多巴胺-1(D1)和/或多巴胺-2(D2)受体的活性。多巴胺D2受体分为两类:D2L和D2S,这是由单个基因通过差别剪接而形成的。D2L受体的胞内域比D2S长。在一些实施方案中,多巴胺受体调节剂抑制配体结合的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%,如在本文所述的试验中所测定的那样。在一些实施方案中,与在相同个体中用多巴胺受体调节剂治疗之前的相应的活性相比,或与在没有接受多巴胺受体调节剂的其他个体中的相应的活性相比,多巴胺受体调节剂降低多巴胺受体的活性的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%。在一些实施方案中,与在相同个体中用多巴胺受体调节剂治疗之前的相应的活性相比,或与在没有接受多巴胺受体调节剂的其他个体中的相应的活性相比,多巴胺受体调节剂增强增强多巴胺受体的活性的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%或200%或300%或400%或500%或更多。在一些实施方案中,多巴胺受体调节剂能够结合于多巴胺受体的活性位点(例如,配体的结合位点)。在一些实施方案中,多巴胺受体调节剂能够结合于多巴胺受体的别构位点。本文使用的术语“血清素受体调节剂”是指且包括结合于或抑制配体结合于血清素受体或者降低或消除或增加或增强或模拟的血清素受体活性的化合物。因此“血清素受体调节剂”包括血清素受体拮抗剂和血清素受体激动剂。在一些实施方案中,血清素受体调节剂以可逆的或不可逆的方式结合于或抑制配体结合于5-HT1A和/或5-HT1B和/或5-HT2A和/或5-HT2B和/或5-HT2C和/或5-HT3和/或5-HT4和/或5-HT6和/或5-HT7受体或者降低或消除或增加或增强或模拟5-HT1A和/或5-HT1B和/或5-HT2A和/或5-HT2B和/或5-HT2C和/或5-HT3和/或5-HT4和/或5-HT6和/或5-HT7受体的活性。在一些实施方案中,血清素受体调节剂抑制配体结合的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%,如在本文所述的试验中所测定的那样。在一些实施方案中,与在相同个体中用血清素受体调节剂治疗之前的相应的活性相比,或与在没有接受血清素受体调节剂的其他个体中的相应的活性相比,血清素受体调节剂降低血清素受体的活性的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%。在一些实施方案中,与在相同个体中用血清素受体调节剂治疗之前的相应的活性相比,或与在没有接受血清素受体调节剂的其他个体中的相应的活性相比,血清素受体调节剂增强血清素受体的活性的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%或200%或300%或400%或500%或更多。在一些实施方案中,血清素受体调节剂能够结合于血清素受体的活性位点(例如,配体的结合位点)。在一些实施方案中,血清素受体调节剂能够结合于血清素受体的别构位点。本文使用的术语“组胺受体调节剂”是指且包括结合于或抑制配体结合于组胺受体或者降低或消除或增加或增强或模拟组胺受体活性的化合物。因此“组胺受体调节剂”包括组胺受体拮抗剂和组胺受体激动剂。在一些实施方案中,组胺受体调节剂以可逆的或不可逆的方式结合于或抑制配体结合于组胺H1和/或H2和/或H3受体或者降低或消除或增加或增强或模拟组胺H1和/或H2和/或H3受体的活性。在一些实施方案中,组胺受体调节剂抑制配体结合的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%,如在本文所述的试验中所测定的那样。在一些实施方案中,与在相同个体中用组胺受体调节剂治疗之前的相应的活性相比,或与在没有接受组胺受体调节剂的其他个体中的相应的活性相比,组胺受体调节剂降低组胺受体的活性的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%。在一些实施方案中,与在相同个体中用组胺受体调节剂治疗之前的相应的活性相比,或与在没有接受组胺受体调节剂的其他个体中的相应的活性相比,组胺受体调节剂增强组胺受体的活性的至少约或约以下各值中的任何一个:10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%或200%或300%或400%或500%或更多。在一些实施方案中,组胺受体调节剂能够结合于组胺受体的活性位点(例如,配体的结合位点)。在一些实施方案中,组胺受体调节剂能够结合于组胺受体的别构位点。除非另外清楚地说明,本文使用的术语“个体”是指哺乳动物,包括但不限于人类、牛类动物(bovine)、灵长类动物、马类动物(equine)、犬科动物(canine)、猫科动物(feline)、猪类动物(porcine)和绵羊类动物(ovine)。因而,本发明可用于人类药物及兽医范畴,其包括用于农业动物和家庭宠物。所述个体可以是已被诊断为或疑似患有认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症的人类。所述个体可以是表现出与认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症有关的一种或多种症状的人类。所述个体可以是具有与认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症有关的突变基因或异常基因的人类。所述个体可以是由于遗传学或其他原因而易于罹患认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症的人类。本文使用的“治疗”是指一种获得有利结果或期望的结果、包括临床结果的方法。本发明中,有利的或期望的临床结果包括但不限于缓解与疾病或病症相关的症状和/或减轻与疾病或病症相关的症状程度和/或预防与疾病或病症相关的症状恶化。在一个实施方案中,有利的或期望的临床结果包括但不限于缓解与认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症相关的症状和/或减轻所述症状程度和/或预防所述症状恶化。优选地,使用本发明化合物或其可药用盐治疗疾病或病症没有副作用或副作用少于与目前可用的对所述疾病或病症的治疗法相关的副作用,和/或改善个体的生活质量。本文使用的“延缓”疾病或病症的发展是指延迟、阻碍、减缓、延缓、稳定和/或推迟所述疾病或病症的发展。这种延缓可以是改变时间长度,其取决于病史和/或接受治疗的个体。对本领域技术人员而言显而易见的是,在未发展为所述疾病或病症的个体中充分的或显著的延缓实际上可以包括预防。例如,“延缓”阿尔茨海默病发展的方法是指与不使用该方法相比,降低在给定时限内疾病发展可能性的方法和/或降低在给定时限内疾病的程度的方法。这种比较通常基于临床研究,使用统计学上具有显著意义的数目的研究对象。例如,使用标准的临床技术诸如常规神经学检查、患者诊视、神经成像、检测血清中或脑脊液中特定蛋白(例如淀粉样肽和Tau)的水平变化、计算机断层摄影术(CT)或核磁共振成像(MRI)可检测到阿尔茨海默病的发展。本领域已知针对其他疾病和病症的类似的技术。发展还指开始时检测不到的疾病进展,且包括出现、复发和发作。本文使用的“有风险”的个体是指有发展为能用本发明化合物治疗的认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症的风险的个体。“有风险”的个体在本文所述治疗方法之前可能罹患或可能未罹患可检测到的疾病或病症,可能表现出或可能未表现出可检测到的疾病。“有风险”是指个体具有一种或多种所谓的风险因素,这些风险因素是与疾病或病症的发展具有相关性的可测量参数且为本领域所知。具有一种或多种这些风险因素的个体与不具有这些风险因素的个体相比,发展为所述疾病或病症的概率更高。这些风险因素包括但不限于年龄、性别、种族、饮食、病史、前体疾病的存在、基因(即遗传)因素和环境暴露。例如有罹患阿尔茨海默病风险的患者包括例如其亲属罹患该病的人和通过基因指标或生化指标分析被确定为有风险的人。患阿尔茨海默病的风险基因指标包括APP基因突变,特别是分别被称为哈迪(Hardy)突变和瑞典(Swedish)突变的717位突变以及670和671位突变(Hardy,TrendsNeurosci.,20:154-9,1997)。其它风险指标有早老素基因突变(例如PS1或PS2)、ApoE4等位基因、阿尔茨海默病家族病史、高胆固醇血症和/或动脉粥样硬化。针对其他疾病和病症的其他的此类因素为本领域所知。本文使用的术语“促-认知(pro-cognitive)”包括但不限于一种或多种心理过程(诸如记忆、注意力、感知和/或思维)的改善,其可通过本领域已知的方法评价。本文使用的术语“神经营养”效应包括但不限于增强神经元功能诸如生长、存活和/或神经递质合成的效应。本文使用的术语“认知障碍”涉及且是指被认为或确实牵涉于神经元结构和/或功能的进行性损失(包括神经元死亡)或与上述有关的疾病和病症,且其中所述障碍的主要特征是认知(例如,记忆、注意力、感知和/或思维)的损伤。这些障碍包括病原体-诱导的认知功能障碍,例如HIV相关的认知功能障碍和莱姆病相关的认知功能障碍。认知障碍的实例包括阿尔茨海默病、亨廷顿病、帕金森病、肌萎缩侧索硬化(ALS)、孤独症、轻度认知功能损害(MCI)、中风、创伤性脑损伤(TBI)和与年龄相关的记忆损害(AAMI)。本文使用的术语“精神病性精神障碍”涉及且是指被认为或确实引起异常思维和感知的精神疾病或病症。精神病性精神障碍以脱离现实为特征,可伴随有妄想、幻觉(是指缺乏外界刺激作用的情况下在有意识和清醒状态的感知,其具有真实的感知的特性,在这种情况下这些感知生动、明显,且位于外部客观空间)、人格改变和/或思维瓦解。其他的常见症状包括反常或古怪的行为,以及社会交往困难和进行日常活动的能力损伤。示例性的精神病性精神障碍为精神分裂症、双相性精神障碍、精神病、焦虑和抑郁。本文使用的术语“神经递质-介导的障碍”涉及且是指被认为或确实牵涉于神经递质诸如组胺、血清素、多巴胺、去甲肾上腺素水平异常或胺能G蛋白偶联受体功能损伤或与上述有关的疾病或病症。示例性的神经递质-介导的障碍包括脊髓损伤、糖尿病性神经病、变应性疾病和牵涉于老化保护(geroprotective)活性的疾病,诸如年龄相关的掉发(脱发)、年龄相关的体重减轻和年龄相关的视觉障碍(白内障)。异常的神经递质水平与众多疾病和病症相关,其包括但不限于阿尔茨海默病、帕金森病、孤独症、吉兰巴雷综合征、轻度认知功能损害、精神分裂症、焦虑、多发性硬化、中风、创伤性脑损伤、脊髓损伤、糖尿病性神经病、纤维肌痛、双相性精神障碍、精神病、抑郁和多种变应性疾病。本文使用的术语“神经元病症”涉及且是指被认为或确实牵涉于神经元细胞死亡和/或神经元功能损伤或神经元功能降低或与上述有关的疾病或病症。示例性的神经元病症包括神经变性疾病和障碍诸如阿尔茨海默病、亨廷顿病、肌萎缩侧索硬化(ALS)、帕金森病、犬认知功能障碍综合征(CCDS)、雷维小体病(Lewybodydisease)、门克斯病、威尔逊病、克雅氏病、法尔病(Fahrdisease)、与脑循环有关的急性或慢性病症诸如缺血性或出血性中风或其它的脑出血性损伤、年龄相关的记忆损害(AAMI)、轻度认知功能损害(MCI)、与创伤有关的轻度认知功能损害(MCI)、脑震荡后综合征、创伤后应激障碍、辅助化学疗法、创伤性脑损伤(TBI)、神经元死亡介导的眼部疾病、黄斑变性、年龄有关的黄斑变性、孤独症(包括孤独症谱系障碍)、阿斯珀格综合征和雷特综合征、撕脱损伤、脊髓损伤、重症肌无力、吉兰巴雷综合征、多发性硬化、糖尿病性神经病、纤维肌痛、与脊髓损伤相关的神经病、精神分裂症、双相性精神障碍、精神病、焦虑或抑郁。本文使用的术语“神经元”表示具有外胚层胚胎起源的细胞,其来源于动物神经系统的任何部分。神经元表达很具有特征性的神经元标记物,包括神经丝蛋白、NeuN(神经元核标记物)、MAP2和III类微管蛋白。作为神经元包括的是例如海马神经元、皮质神经元、中脑多巴胺能神经元、脊髓运动神经元、感觉神经元、交感神经元、中隔胆碱能神经元和小脑神经元。本文使用的术语“神经突向外生长”或“神经突活化”是指已存在的神经元突起(例如,轴突和树突)的延伸和新的神经元突起(例如,轴突和树突)的生长或萌发。神经突向外生长或神经突活化可改变神经连接性,导致建立新的突触或重塑已存在的突触。本文使用的术语“神经发生”是指从未分化的神经元祖细胞产生新的神经细胞,所述神经祖细胞也称为全能神经干细胞。神经发生积极地产生新的神经元、星形细胞、神经胶质、神经膜细胞、少突胶质细胞和/或其他神经谱系。虽然神经发生持续至生命后期,但其大量出现在人类发育的早期,特别是在成人脑中的某些局限的区域。本文使用的术语“神经连接性”是指有机体中神经元之间的连接(“突触”)的数目、类型和性质。突触在神经元之间、在神经元和肌肉之间(“神经肌肉连接头”)以及神经元和其他生物结构(包括器官、内分泌腺等)之间形成。突触具有特定的结构,神经元通过突触向彼此和非神经元细胞、肌肉、组织和器官传递化学或电学信号。影响神经连接性的化合物通过建立新的突触(例如,通过神经突向外生长或神经突活化)或通过改变或重塑已存在的突触可起到这样的作用。突触的重塑是指在特定突触传递的信号的性质、强度或类型的改变。本文使用的术语“神经病”是指特征为神经系统的运动、感觉和自主神经元的功能和/或结构改变的病症,其由神经系统的原发病灶或其他功能障碍引发或导致。周围神经病的类型包括多神经病、单神经病、多发性单神经炎和自主神经病。最常见的形式是(对称)多发性周围神经病,其主要影响脚和腿。神经根病牵涉脊神经根,但是如果还牵涉周围神经则使用术语神经根神经病。神经病的形式可由原因或牵涉的主要纤维的大小而进一步细分,例如大纤维或小纤维周围神经病。中枢神经病性疼痛可出现于脊髓损伤、多发性硬化和某些中风以及纤维肌痛。神经病可伴随虚弱、自主性变化和感觉变化的不同组合。也可看到肌肉块的丧失或肌束自发性收缩、一种特定的细微的肌肉颤搐。感觉性症状包括感觉和“正性”现象(包括疼痛)的丧失。神经病与多种病症相关,包括糖尿病(例如,糖尿病性神经病)、纤维肌痛、多发性硬化和带状疱疹感染以及脊髓损伤和其他类型的神经损伤。本文使用的术语“阿尔茨海默病”是指变性的脑部疾病,其临床特征是进行性记忆缺失、意识错乱、行为问题、不能自理、逐步的体质恶化并最终死亡。该疾病在组织学上的特征是神经炎性斑块,其主要发现在联络皮质、边缘系统和基底神经节。这些斑块的主要组成是淀粉样β肽(Aβ),其是β淀粉样前蛋白的裂解产物(βAPP或APP)。APP是I型跨膜糖蛋白,其包含一个大的异位N-末端区域、一个跨膜区和一个小的细胞质C-末端尾部。选择性剪接染色体21上的单个APP基因的转录物产生若干氨基酸数目不同的同工型。Aβ显示在阿尔茨海默病的神经病理学中具有主要的作用。已证明该疾病的家族型与APP和早老素基因中的突变的联系(Tanzi等,1996,Neurobiol.Dis.,3:159-168;Hardy,1996,Ann.Med.,28:255-258)。在这些基因中的与疾病相关的突变导致Aβ的42-氨基酸形式的产生增加,其是在淀粉样斑块中发现的主要形式。还已报道线粒体功能障碍是阿尔茨海默病的重要成分(Bubber等,MitochondrialabnormalitiesinAlzheimerbrain:MechanisticImplications(在阿尔茨海默脑中的线粒体异常:机理推断),AnnNeurol,2005,57(5),695-703;Wang等,Insightsintoamyloid-β-inducedmitochondrialdysfunctioninAlzheimerdisease(对在阿尔茨海默病中淀粉样蛋白β诱导的线粒体功能障碍的研究),FreeRadicalBiology&Medicine,2007,43,1569-1573;Swerdlow等,MitochondriainAlzheimer’sdisease(阿尔茨海默病中的线粒体),Int.Rev.Neurobiol.,2002,53,341-385;和Reddy等,AremitochondriacriticalinthepathogenesisofAlzheimer’sdisease?(线粒体在阿尔茨海默病中是关键性的吗?),BrainResRev.2005,49(3),618-32)。已提出线粒体功能障碍是神经元功能(包括神经递质合成和分泌)和活力的原因。因此,稳定线粒体的化合物可对阿尔茨海默病患者具有有益的作用。本文使用的术语“亨廷顿病”是指致命的神经病学疾病,其临床特性是诸如不自主的运动、认知损伤或丧失认知功能和广谱的行为障碍的症状。亨廷顿病伴随的常见运动症状包括舞蹈症(非自主的扭动和痉挛)、笨拙和走路、说话(例如,表现出言语不清)和吞咽能力的进行性丧失。其他亨廷顿病症状可包括认知症状诸如用脑速度、注意力和短期记忆丧失和/或行为症状,所述行为症状可包括性格改变、抑郁、易激惹、情感爆发和情感淡漠症。临床症状通常出现在生命的第四或第五个十年。亨廷顿病是破坏性的且通常长期的疾病,通常在症状出现后约10-20年死亡。亨廷顿病通过编码称为突变亨廷顿蛋白(mutanthuntingtinprotein)的异常蛋白的突变的或异常的基因而遗传;突变亨廷顿蛋白造成许多不同的脑区域中神经元变性。所述变性集中在位于基底神经节的神经元、在脑的深部控制许多重要功能(包括协调活动)的结构、以及在脑或皮质外表面上的神经元,其控制思维、感知和记忆。本文所用的“肌萎缩侧索硬化”或“ALS”表示进行性神经变性疾病,其侵袭上位运动神经元(脑中的运动神经元)和/或下位运动神经元(脊髓中的运动神经元)并导致运动神经元死亡。本文所用的术语“ALS”包括本领域中已知的ALS的所有分类,其包括但不限于典型的ALS(通常侵袭下位和上位运动神经元)、原发性侧索硬化(PLS,通常仅侵袭上位运动神经元)、进行性延髓麻痹(PBP或延髓发作,其是ALS的一种型式,其通常始于吞咽、咀嚼和说话困难)、进行性肌萎缩(PMA,通常仅侵袭下为运动神经元)和家族性ALS(一种遗传性的ALS型式)。本文使用的术语“帕金森病”是指其中个体感受到一种或多种与帕金森病相关的症状的任何医学病症,诸如(非限制性)以下一种或多种症状:休息性震颤、齿轮样强直、运动迟缓、姿势反射损伤、对1-多巴治疗具有良好响应的症状、没有显著的动眼神经麻痹、小脑或锥体束征、肌萎缩、运动障碍和/或言语障碍。在一个特定的实施方案中,本发明用于治疗多巴胺能功能障碍相关的病症。在一个特定的实施方案中,患有帕金森病的个体在突触核蛋白、parkin或NURR1核酸(其与帕金森病相关)中具有突变或多态性。在一个实施方案中,患有帕金森病的个体核酸的表达有缺陷或降低或核酸有突变,所述核酸调节调节多巴胺能神经元的发育和/或存活。本文使用的术语“犬认知功能障碍综合征”或“CCDS”是指年龄相关的精神功能恶化,其特征是影响患病的犬科动物正常发挥功能的能力的多重认知损伤。与CCDS相关的认知能力的降低不能完全归因于通常的医学病症诸如瘤形成、感染、感觉损伤或器官衰竭。犬科动物诸如狗中CCDS的诊断通常是排除的诊断,其基于彻底的行为和医疗史以及与其他疾病过程无关的CCDS临床症状的存在。主人对在行为上与年龄相关的变化的观察是用于发现年老的家养狗中可能的CCDS发作的实用方法。可使用许多实验室的认知任务来帮助诊断CCDS,同时可以使用血细胞技术、化学仪器和尿液分析来排除其他可能与CCDS的临床症状很像的潜在疾病。CCDS的症状包括记忆损失(在家养狗中其可由定向障碍和/或意识错乱而证明)、与家庭成员之间的互动和/或问候行为减少或改变、睡醒循环改变、活动水平降低和家庭训练丧失或频繁的、不适当的排泄。患CCDS犬科动物可显示出一种或多种以下的临床或行为症状:食欲降低、对环境的察觉降低、识别熟悉位置、人或其他动物的能力降低、听力减退、上下楼梯的能力降低、对孤独的耐受性降低、强迫行为或重复行为或习惯的发展、转圈、震颤或摇动、定向障碍、活动水平降低、睡醒循环异常、家庭训练丧失、对家庭成员的反应性降低或改变、以及问候行为的降低或改变。CCDS可显著影响患病的犬科动物的健康和快乐。而且,当CCDS加重和其症状变得更加严重时,由患该疾病的宠物带来的伙伴关系的益处会变得更少。本文使用的术语“年龄相关的记忆损害”或“AAMI”是指可鉴定为整体衰退量表(GDS)上的GDS2期的病症(Reisberg等.(1982)Am.J.Psychiatry139:1136-1139),GDS将衰老过程和进行性变性痴呆分为七个主要的阶段。GDS的第一期是其中在任何年龄的个体既没有认知损害的主诉也没有损害的客观证据的阶段。认为这些GDS1期患者是正常的。GDS的第二期适用于那些通常年龄较大的人,其抱怨记忆和认知功能的困难,诸如想不起名字,如同其在五或十年前那样,或者想不起他们把东西放在何处,如同其在五或十年前那样。这些主诉在其他正常的老人中显得非常常见。AAMI是指GDS2期的人,其在神经生理学上可不同于正常且没有主诉的老人(即GDS1期)。例如,已发现,AAMI个体在计算机分析的EEG上相比GDS1期老人在电生理上更慢(Prichep,John,Ferris,Reisberg等.(1994)Neurobiol.Aging15:85-90)。本文使用的术语“轻度认知功能损害”或“MCI”是指一种类型的认知障碍,其特征是认知功能比对正常的与年龄相关的衰退而言通常所显示的那样更加显著的恶化。因此,患MCI的老人或年龄大的人在进行复杂的日常工作和学习时比正常情况有更大的困难,但不存在阿尔茨海默病患者中典型的不能进行正常的日常社交和/或专业职能,或最终导致痴呆的其他相似的神经变性病症。MCI的特征是轻微的、临床上明显的在认知、记忆和发挥功能中的缺陷等损害,其级别不足以符合阿尔茨海默病或其他痴呆的诊断标准。MCI还包括损伤相关的MCI,所述损伤相关的MCI在本文定义为由某些类型的损伤导致的认知损害,诸如神经损伤(即战场损伤,包括脑震荡后综合征等)、神经毒性的治疗(即,导致“化疗脑”的助化学疗法等)和由于物理性损伤或其他神经变性引起的组织损伤,其独立于且不同于由中风、缺血、出血性损伤、钝性力量创伤等导致的轻度认知功能损害。本文使用的术语“创伤性脑损伤”或“TBI”是指由突发性创伤导致的脑损伤,诸如击打或震摇或穿通性头部外伤,其破坏脑功能或破坏大脑。TBI的症状的范围可以是从轻度、中度至重度,且可显著影响认知的(语言和交流、信息加工、记忆和感知技能缺陷)、身体的(离床活动、平衡、协调、精细运动、力量和忍耐性)和心理学技能。“神经元死亡介导的眼部疾病”是指眼部疾病,其中在整体上或部分牵涉神经元的死亡。所述疾病可涉及光感受器的死亡。所述疾病可涉及视网膜细胞的死亡。所述疾病可涉及眼神经通过细胞凋亡而死亡。具体的神经元死亡介导的眼部疾病包括但不限于黄斑变性、青光眼、色素性视网膜炎、先天性静止性夜盲症(小口氏病)、儿童期发作的严重视网膜营养性萎缩、莱伯先天性黑朦、巴-比综合征、厄舍尔综合征、视神经病变导致的失明、莱伯遗传性视神经病、色盲和Hansen-Larson-Berg综合征。本文使用的术语“黄斑变性”包括本领域已知的所有形式和分类的黄斑变性,其包括但不限于特征为与布鲁赫膜、脉络膜、神经视网膜和/或视网膜色素上皮异常有关的进行性中心视力丧失的疾病。因此该术语包括诸如年龄相关性黄斑变性(ARMD)以及罕见的较早发病的营养不良(在一些情况下其可在生命的首个十年中被检测到)的病症。其他黄斑病变包括北卡罗莱纳黄斑营养不良、索斯比眼底营养不良、施塔加病、图形样营养不良(patterndystrophy)、贝斯特病和MalattiaLeventinese。本文使用的术语“孤独症”是指一种脑发育障碍,其损害社会交往和沟通并导致有限的和重复的行为,通常出现在婴儿期或童年早期。认为该认知和行为缺陷是部分由神经连接性改变而导致的。孤独症包括有时称为“孤独症谱系障碍”的相关的障碍以及阿斯珀格综合征和雷特综合征。本文使用的术语“神经损伤”是指对神经的物理性损伤,诸如撕脱损伤(即,神经已被撕裂或拉开之处)或脊髓损伤(即,白质或向脑传送感觉和运动信号和从脑传出感觉和运动信号的有髓鞘神经纤维束的损伤)。许多原因可导致脊髓损伤的出现,包括物理性创伤(即,车祸、运动损伤等)、侵犯脊柱的肿瘤、发育障碍,诸如脊柱裂等。本文使用的术语“重症肌无力”或“MG”是指非认知性的神经肌肉障碍,其是由免疫介导的骨骼肌的神经肌肉接头的乙酰胆碱受体的损失导致的。临床上,在约三分之二的患者中MG通常首先以偶然的肌无力出现,在眼外肌中最常见。这些最初的症状最终恶化,造成眼睑下垂(上睑下垂症)和/或复视,常常使得患者去寻求医疗帮助。最后,许多患者发展成全身性肌无力,其可能每周、每日或甚至更频繁地波动。全身性的MG常常累及控制面部表情、咀嚼、说话、吞咽和呼吸的肌肉;在近期的治疗进展之前,呼吸衰竭是最常见的死因。本文使用的术语“吉兰巴雷综合征”是指非认知障碍,其中身体的免疫系统攻击部分周围神经系统。该病症首先的症状包括腿部不同程度的无力或刺痛感。在许多情况下所述无力和刺痛感扩散至胳膊和上身。这些症状可在强度上增加,直至某些肌肉完全不能使用,且当严重时,患者几乎完全瘫痪。在这些情况下所述病症是威胁生命的——潜在地干扰呼吸且有时干扰血压或心率,并且认为所述疾病在医学上是紧急的。然而大部分患者从甚至最严重的吉兰巴雷综合征中得到恢复,虽然有些继续具有某种程度的无力。本文使用的术语“多发性硬化”或“MS”是指自身免疫病症,其中免疫系统攻击中枢神经系统(CNS),导致神经元脱髓鞘。其可导致多种症状,其中许多是非认知性的,且常常发展为身体残疾。MS侵袭称为白质的脑和脊髓的区域。当完成加工处理时,白质细胞在灰质区域和身体的其余部分之间传送信号。更具体而言,MS破坏少突胶质细胞,其是承担创建和维持脂肪层(称为髓鞘)作用的细胞,髓鞘帮助神经元携带电信号。MS导致薄的或完全丧失的髓磷脂,并且偶尔切断(横断)神经元的延长部分或轴突。当丢失髓磷脂时,神经元不再能有效地传导其电信号。所述疾病可伴随几乎任何神经病学症状。MS呈现若干种形式,其中新的症状以不连续的发作(复发的形式)出现或随时间慢慢累积(进行性的形式)而出现。大部分人首先被诊断具有复发-缓解型MS,但在数年后发展为继发进行性MS(SPMS)。在各次发作之间,症状可能完全消失,但是持久的神经病学问题常常持续存在,尤其当疾病进展时。本文使用的术语“精神分裂症”是指慢性的精神障碍,其特征是一种或多种正性症状(例如,妄想和幻觉)和/或负性症状(例如,情感迟滞和缺乏兴趣)和/或混乱症状(例如,思维和语言瓦解或感知和行为混乱)。本文所使用的精神分裂症包括本领域中已知的精神分裂症的所有形式和分类,其包括但不限于紧张型、青春型、混乱型、偏执型、残余型或未定型精神分裂症和缺陷综合征和/或在美国精神病协会:DiagnosticandStatisticalManualofMentalDisorders(精神障碍的诊断和统计手册),第四版,华盛顿,2000或在InternationalStatisticalClassificationofDiseasesandRelatedHealthProblems(疾病和相关健康问题的国际统计分类)中所描述的那些,或其他本领域技术人员已知的那些。本文使用的术语“老化保护活性”或“老化保护剂”表示通过降低并不威胁生命但与老化过程有关且对老人而言是典型的病理或状况的量和/或强度的水平,来减慢老化和/或延长生命和/或提高或改善生命质量的生物活性。并不威胁生命但与老化过程有关的病理或状况包括这样的病理或状况,如失明(白内障)、有皮肤毛发的皮肤(dermatohairyintegument)的恶化(脱发)和由于肌肉和/或脂肪细胞的死亡导致的年龄相关的体重降低。本文使用的术语“变应性疾病”是指免疫系统的病症,其特征是过度活化肥大细胞和嗜碱性细胞和产生IgE免疫球蛋白,导致强烈的免疫应答。其代表了对称为变应原的环境物质的超敏反应的一种形式,且是获得型疾病。常见的变应性反应包括湿疹、荨麻疹、枯草热、哮喘、食物过敏、对蛰刺昆虫诸如黄蜂和蜜蜂的毒液的反应。变应性反应伴随着组胺的过度释放,且因此可用抗组胺剂来治疗。用于本文的术语“联合治疗”是指包括两种或更多种不同的化合物的治疗。因而,一方面提供了包含本文详述的化合物和另一个化合物的联合治疗。在一些实施方案中,联合治疗任选地包括一种或多种可药用载体或赋形剂、非药物活性化合物和/或惰性物质。在多个实施方案中,与施用单独的本发明化合物相比采用联合治疗的治疗方案可产生加合的或甚至是协同的(例如,大于加合)效果。在一些实施方案中,与各化合物单独治疗通常使用的量相比,每一个化合物作为联合治疗的一部分而使用的量较少。优选地,使用联合治疗与单独使用单个的化合物中的任何一个相比获得相同的或更大的治疗益处。在一些实施方案中,与单独的化合物或单独治疗通常使用的量相比,在联合治疗中使用较少的量(例如,较低的剂量或较低频率的给药方案)的化合物会达到相同的或更大治疗益处。优选地,较少剂量的化合物的使用导致减少该化合物伴随的一种或多种副作用的数量、严重性、频率和/或持续时间。术语“有效量”是指结合其功效和毒性参数并且根据执业专业人员的知识判断在给定的治疗形式中应该是有效的本发明化合物的量。根据现有技术的理解,有效量可以是一个或多个剂量,即获得期望的治疗终点可能需要单剂量或多剂量。有效量可以放在施用一种或多种治疗剂的情况中考虑,而且如果与一种或多种其它治疗剂组合可能获得或获得期望结果或有利结果则可认为单个治疗剂是有效量的。由于各化合物之间产生联合的作用(例如加合作用或协同作用),共同施用的任一化合物的合适剂量可任选地减少。用于本文的“单位剂型”是指适合作为单位剂量的物理上分离的单位,每个单位包含计算出的用来产生需要的治疗作用的预定量的活性成分以及所需的药用载体。单位剂型可包含单个或联合治疗。本文使用的术语“控释”是指药物不被立即释放的含药制剂或它的一部分,即“控释”制剂的施用不会导致药物立即释放至吸收池中。该术语包括设计用来在延长的时间段内逐步释放药物化合物的储库制剂。控释制剂能包括众多药物释放系统,通常涉及将药物化合物与具有所需释放特性(例如,pH-依赖性或非-pH-依赖溶解度、水溶性的不同程度等)的载体、聚合物或其他化合物混合,并依据所需递送途径(例如,包衣胶囊、可植入贮库、含可降解胶囊的注射溶液等)配制该混合物。用于本文的“可药用”或“药理学可接受”是指在生物学或在其它方面不属于不期望的材料,例如所述材料可以掺入施用于患者的药物组合物中而不会引起任何显著的不期望的生物学后果或与该组合物中任何其它组分发生有害相互作用。可药用载体或赋形剂优选已满足毒理学检测和制药检测要求的标准,和/或收录于美国FDA编撰的《惰性成分指南》中。“可药用盐”是那些保留了游离(非盐)化合物的至少一些生物活性的且能作为药物或药物制剂施用于个体的盐。可药用盐是指离子相互作用而不是共价键。由此N-氧化合物不被认为是盐,例如,包括:(1)酸加成盐,与无机酸形成,所述无机酸诸如盐酸、氢溴酸、硫酸、硝酸、磷酸等;或与有机酸形成,所述有机酸诸如乙酸、草酸、丙酸、琥珀酸、马来酸、酒石酸等;(2)当本发明化合物中的酸性质子被金属离子代替所形成的盐,所述金属离子例如,碱金属离子、碱土金属离子或铝离子;或与有机碱配位所形成的盐。可接受的有机碱包括乙醇胺、二乙醇胺、三乙醇胺等。可接受的无机碱包括氢氧化铝、氢氧化钙、氢氧化钾、碳酸钠、氢氧化钠等。可药用盐进一步的实例包括在Berge等,PharmaceuticalSalts,J.Pharm.ScL1977年1月;66(1):1-19中所列出的那些。可药用盐能在制备过程中在原位制备,或通过单独地分别将纯化的游离酸或游离碱形式的本发明化合物分别与适合的有机或无机碱或酸反应,并在接着的纯化步骤中将由此形成的盐分离。应理解提及的可药用盐包括溶剂加成形式或其结晶形式,特别是溶剂化物或多晶型物。溶剂化物包含化学计量或非化学计量的量的溶剂,且其经常在结晶过程中形成。当溶剂为水时形成水合物,或当溶剂是醇是形成醇化物。多晶型物包括一个化合物的相同元素组成的不同的晶体堆积排列。多晶型物通常具有不同的X-射线衍射图谱、红外光谱、熔点、密度、硬度、晶形、光学或电学性质、稳定性和溶解度。多种因素诸如重结晶溶剂、结晶速度和贮存温度可引起单个晶形占优势。用于本文的术语“赋形剂”是指可用于生产药物或药剂(诸如包含作为活性成分的本发明化合物的片剂)的惰性或无活性物质。术语赋形剂可包括多种物质,其非限制性的包括用作粘合剂、崩解剂、包衣、压缩/包囊助剂、霜剂或洗剂、润滑剂、用于胃肠外施用的溶液、用于咀嚼片的物质、甜味剂或矫味剂、助悬剂/胶凝剂或湿法制粒剂的任何物质。粘合剂包括例如,卡波姆、聚维酮、黄原胶等;包衣包括例如,醋酞纤维素、乙基纤维素、结冷胶、麦芽糖糊精、肠溶衣等;压缩/包囊助剂包括例如,碳酸钙、右旋糖、果糖dc(dc=“直接可压缩”)、蜂蜜dc、乳糖(无水或一水合物;任选地与阿司帕坦、纤维素或微晶纤维素组合)、淀粉dc、蔗糖等;崩解剂包括例如,交联羧甲纤维素钠、结冷胶、羟乙酸淀粉钠等;霜剂或洗剂包括例如,麦芽糖糊精、角叉菜胶等;润滑剂包括例如,硬脂酸镁、硬脂酸、硬脂酰醇富马酸钠等;用于咀嚼片的物质包括例如,右旋糖、果糖dc、乳糖(一水合物、任选地阿司帕坦或纤维素组合)等;助悬剂/胶凝剂包括例如,角叉菜胶、羧羟乙酸淀粉钠、黄原胶等;甜味剂包括例如,阿司帕坦、右旋糖、果糖dc、山梨醇、蔗糖dc等;且湿法制粒剂包括例如,碳酸钙、麦芽糖糊精、微晶纤维素等。“烷基”是指且包括饱和的直链、支链或环状一价烃结构及其组合。具体的烷基是具有1至20个碳原子(“C1-C20烷基”)的那些烷基。更具体的烷基是具有1至8个碳原子(“C1-C8烷基”)的那些烷基。当提及具有特定数目的碳的烷基时,意在包括和描述所有具有该数目碳的几何异构体;因而,例如,“丁基”意在包括正-丁基、仲-丁基、异-丁基、叔-丁基和环丁基;“丙基”包括正-丙基、异-丙基和环丙基。该术语通过以下基团举例说明:诸如甲基、叔-丁基、正-庚基、辛基、环己基甲基、环丙基等。环烷基是烷基的一个亚组且能形成一个环,诸如环己基或多个环诸如金刚烷基。包含超过一个环的环烷基可以是稠合的、螺状的或桥联的或是其组合。在稠合环系统中,一个或多个环可以是芳基或杂芳基。具有超过一个的环且其中至少一个环是芳环的环烷基可在非芳环位置或在芳环位置上连接于母体结构。在一个实施方案中,具有超过一个的环且其中至少一个环是芳环的环烷基在非芳环位置上连接于母体结构。优选的环烷基是具有3至13个环碳原子的饱和环烃。更优选的环烷基是具有3至7个环碳原子(“C3-C7环烷基”)的饱和环烃。环烷基的实例包括金刚烷基、十氢萘基、环丙基、环丁基、环戊基等。“亚烷基”是指与烷基相同的基团,但具有二价。亚烷基的实例包括亚乙基(-CH2CH2-)和亚丙基(-CH2CH2CH2-)。“链烯基”是指具有至少一个烯属不饱和位点(即,具有至少一个式C=C基团)且优选具有2至10个碳原子、更优选具有2至8个碳原子的不饱和烃基团。链烯基的实例包括但不限于-CH2-CH=CH-CH3和-CH2-CH2-环己烯基,其中后者的乙基能在环上任何可用的位置上连接于环己烯基。“炔基”是指具有至少一个炔属不饱和位点(即,具有至少一个式C≡C基团)且优选具有2至10个碳原子、更优选具有3至8个碳原子的不饱和的烃基团。“被取代的烷基”是指具有1至5个取代基的烷基,所述取代基包括但不限于取代基诸如烷氧基、被取代的烷氧基、酰基、酰氧基、羰基烷氧基、酰基氨基、被取代的或未被取代的氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、芳氧基、被取代的芳氧基、氰基、卤素、羟基、硝基、羧基、硫羟、烷硫基、被取代的或未被取代的链烯基、被取代的或未被取代的炔基、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、氨基磺酰基、磺酰基氨基、磺酰基、氧代、羰基亚烷基烷氧基等。“被取代的链烯基”是指具有1至5个取代基的链烯基,所述取代基包括但不限于取代基诸如烷氧基、被取代的烷氧基、酰基、酰氧基、羰基烷氧基、酰基氨基、被取代的或未被取代的氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、芳氧基、被取代的芳氧基、氰基、卤素、羟基、硝基、羧基、硫羟、烷硫基、被取代的或未被取代的烷基、被取代的或未被取代的炔基、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、氨基磺酰基、磺酰基氨基、磺酰基、氧代、羰基亚烷基烷氧基等。“被取代的炔基”是指具有1至5个取代基的炔基,所述取代基包括但不限于基团诸如烷氧基、被取代的烷氧基、酰基、酰氧基、羰基烷氧基、酰基氨基、被取代的或未被取代的氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、芳氧基、被取代的芳氧基、氰基、卤素、羟基、硝基、羧基、硫羟、烷硫基、被取代的或未被取代的烷基、被取代的或未被取代的链烯基、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、氨基磺酰基、磺酰基氨基、磺酰基、氧代、羰基亚烷基烷氧基等。“酰基”是指H-C(O)-、烷基-C(O)-、被取代的烷基-C(O)-、链烯基-C(O)-、被取代的链烯基-C(O)-、炔基-C(O)-、被取代的炔基-C(O)-、芳基-C(O)-、被取代的芳基-C(O)-、杂芳基-C(O)-、被取代的杂芳基-C(O)-、杂环-C(O)-和被取代的杂环-C(O)-、其中烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、环烷基、被取代的环烷基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环如本文所定义。“酰氧基”是指H-C(O)O-、烷基-C(O)O-、被取代的烷基-C(O)O-、链烯基-C(O)O-、被取代的链烯基-C(O)O-、炔基-C(O)O-、被取代的炔基-C(O)O-、芳基-C(O)O-、被取代的芳基-C(O)O-、杂芳基-C(O)O-、被取代的杂芳基-C(O)O-、杂环-C(O)O-和被取代的杂环-C(O)O-、其中烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、环烷基、被取代的环烷基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环如本文所定义。“杂环”“杂环的”或“杂环基”是指具有单个环或多个稠合的环且具有1至10个环碳原子和1至4个环杂原子诸如氮、硫或氧的饱和的或不饱和的非芳族基团。包含超过一个环的杂环可以是稠合的、螺状的或桥联的或是其任何组合。在稠合环系统中,一个或多个环可以是芳基或杂芳基。具有超过一个环且其中至少一个环是芳环的杂环可在非芳环位置或在芳环位置上连接于母体结构。在一个实施方案中,具有超过一个环且其中至少一个环是芳环的杂环在非芳环位置上连接于母体结构。“被取代的杂环”或“被取代的杂环基”是指被1至3个取代基所取代的杂环基团,所述取代基包括但不限于取代基诸如烷氧基、被取代的烷氧基、酰基、酰氧基、羰基烷氧基、酰基氨基、被取代的或未被取代的氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、芳氧基、被取代的芳氧基、氰基、卤素、羟基、硝基、羧基、硫羟、烷硫基、被取代的或未被取代的烷基、被取代的或未被取代的链烯基、被取代的或未被取代的炔基、被取代的或未被取代的芳烷基、氨基磺酰基、磺酰基氨基、磺酰基、氧代、羰基亚烷基烷氧基等。在一个实施方案中,被取代的杂环是被另外的环取代的杂环,其中所述另外的环可以是芳族的或非芳族的。“芳基”或“Ar”是指具有单个环(例如,苯基)或多个稠合的环(例如,萘基或蒽基)的不饱和的芳族碳环基团,其中稠合的环可能是或可能不是芳族的。在一个实施方案中,芳基包含6至14个环碳原子。具有超过一个环且其中至少一个环是非芳环的芳基可在芳环位置或在非芳环位置上连接于母体结构。在一个实施方案中,具有超过一个环且其中至少一个环是非芳环的芳基在芳环位置上连接于母体结构。“杂芳基”或“HetAr”是指具有2至10个环碳原子和至少一个环杂原子的不饱和的芳族碳环基团,所述杂原子包括但不限于诸如氮、氧和硫的杂原子。杂芳基可具有单个环(例如,吡啶基、呋喃基)或多个稠合的环(例如,中氮茚基、苯并噻吩基),其中稠合的环可能是或可能不是芳族的。具有超过一个环且其中至少一个是非芳环的杂芳基可在芳环位置或在非芳环位置上连接于母体结构。在一个实施方案中,具有超过一个环且其中至少一个环是非芳环的杂芳基在芳环位置上连接于母体结构。“被取代的芳基”是指具有1至5个取代基的芳基,所述取代基包括但不限于基团诸如烷氧基、被取代的烷氧基、酰基、酰氧基、羰基烷氧基、酰基氨基、被取代的或未被取代的氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、芳氧基、被取代的芳氧基、氰基、卤素、羟基、硝基、羧基、硫羟、烷硫基、被取代的或未被取代的烷基、被取代的或未被取代的链烯基、被取代的或未被取代的炔基、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、氨基磺酰基、磺酰基氨基、磺酰基、氧代、羰基亚烷基烷氧基等。“被取代的杂芳基”是指具有1至5个取代基的杂芳基,所述取代基包括但不限于基团诸如烷氧基、被取代的烷氧基、酰基、酰氧基、羰基烷氧基、酰基氨基、被取代的或未被取代的氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、芳氧基、被取代的芳氧基、氰基、卤素、羟基、硝基、羧基、硫羟、烷硫基、被取代的或未被取代的烷基、被取代的或未被取代的链烯基、被取代的或未被取代的炔基、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、氨基磺酰基、磺酰基氨基、磺酰基、氧代、羰基亚烷基烷氧基、等等(例如,氧化物,诸如当环氮被氧化时)。“芳烷基”是指其中芳基部分连接于烷基上的基团,且其中芳烷基可在芳基或烷基上连接于母体结构。优选地,芳烷基经烷基连接于母体结构。“被取代的芳烷基”是指其中芳基部分连接于被取代的烷基上的基团,且其中芳烷基可在芳基或烷基上连接于母体结构。“烷氧基”是指烷基-O-,其包括,例如甲氧基、乙氧基、正-丙氧基、异-丙氧基、正-丁氧基、叔-丁氧基、仲-丁氧基、正-戊氧基、正-己氧基、1,2-二甲基丁氧基等。相似地,链烯基氧基是指“链烯基-O-”且炔基氧基是指“炔基-O-”。“被取代的烷氧基”是指被取代的烷基-O。“未被取代的氨基”是指-NH2。“被取代的氨基”是指-NRaRb,其中(a)Ra和Rb基团各自独立地选自:H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,条件是Ra和Rb基团不都是H;或(b)Ra和Rb与氮原子一起形成杂环或被取代的杂环。“酰基氨基”是指-C(O)NRaRb,其中Ra和Rb独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环或Ra和Rb基团能与氮原子一起形成杂环或被取代的杂环。“氨基酰基”是指-NRaC(O)Rb,其中Ra和Rb基团各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环。优选地,Ra是H或烷基。“氨基磺酰基”是指-NRSO2-烷基、-NRSO2-被取代的烷基、-NRSO2-链烯基、-NRSO2-被取代的链烯基、-NRSO2-炔基、-NRSO2-被取代的炔基、-NRSO2-芳基、-NRSO2-被取代的芳基、-NRSO2-杂芳基、-NRSO2-被取代的杂芳基、-NRSO2-杂环和-NRSO2-被取代的杂环,其中R是H或烷基,且其中烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、环烷基、被取代的环烷基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环如本文所定义。“磺酰基氨基”是指-SO2NH2、-SO2NR-烷基、-SO2NR-被取代的烷基、-SO2NR-链烯基、-SO2NR-被取代的链烯基、-SO2NR-炔基、-SO2NR-被取代的炔基、-SO2NR-芳基、-SO2NR-被取代的芳基、-SO2NR-杂芳基、-SO2NR-被取代的杂芳基、-SO2NR-杂环和-SO2NR-被取代的杂环,其中R是H或烷基,或-SO2NR2,其中两个R基团与它们所连接的氮原子一起形成杂环或被取代的杂环。“磺酰基”是指-SO2-烷基、-SO2-被取代的烷基、-SO2-链烯基、-SO2-被取代的链烯基、-SO2-炔基、-SO2-被取代的炔基、-SO2-芳基、-SO2-被取代的芳基、-SO2-杂芳基、-SO2-被取代的杂芳基、-SO2-杂环和-SO2-被取代的杂环。“羰基亚烷基烷氧基”是指-C(=O)-(CH2)n-OR,其中R是被取代的或未被取代的烷基,且n是1至100的整数,更优选地n是1至10或1至5的整数。“卤代”或“卤素”是指原子序数为9至85的第17族的元素。优选的卤素基团包括氟、氯、溴和碘基团。当基团被超过一个卤素取代,可使用相应于所连接的卤素数目的前缀来描述所述基团,例如,二卤代芳基、二卤代烷基、三卤代芳基等,就被两个(“二”)或三个(“三”)卤素基团取代的芳基和烷基而言,其可以是但并非必须是相同的卤素;因而4-氯-3-氟苯基在二卤代芳基的范围内。其中每个H均被卤素替代的烷基被称为“全卤代烷基”。优选的全卤代烷基是三氟烷基(-CF3)。相似地,“全卤代烷氧基”是指其中构成烷氧基的烷基部分的烃中的每个H均被卤素取代的烷氧基。全卤代烷氧基的实例是三氟甲氧基(-OCF3)。“羰基”是指C=O。“羧基”是指-C(O)OH。“氰基”是指-CN。“氧代”是指=O。“硝基”是指-NO2。“烷硫基”是指-S-烷基。“烷基磺酰基氨基”是指-R1SO2NRaRb,其中Ra和Rb独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,或Ra和Rb与氮原子一起形成杂环或被取代的杂环,且R1是烷基。“羰基烷氧基”用于本文是指-C(O)O-烷基、-C(O)O-被取代的烷基、-C(O)O-芳基、-C(O)O-被取代的芳基、-C(O)O-链烯基、-C(O)O-被取代的链烯基、-C(O)O-炔基、-C(O)O-被取代的炔基、-C(O)O-杂芳基、-C(O)O-被取代的杂芳基、-C(O)O-杂环或-C(O)O-被取代的杂环。“孪位”是指连接于同一原子的两个基团的关系,例如,在基团-CH2-CHR1R2中,R1和R2是孪位的且R1可被称为R2的孪位R基团。“邻位”是指连接于相邻的原子上的两个基团之间的关系。例如,在基团–CHR1-CH2R2中,R1和R2是邻位的且R1可被称为R2的邻位R基团。“基本上纯的”化合物的组合物是指该组合物包含不超过15%或优选不超过10%或更优选不超过5%或甚至更优选不超过3%和最优选不超过1%的杂质,所述杂质可能是不同立体化学形式的该化合物。例如,基本上纯的S形式的化合物的组合物是指该组合物包含不超过15%或不超过10%或不超过5%或不超过3%或不超过1%的R形式的该化合物。本发明的化合物依据本发明的化合物在本文(包括发明概述和所附权利要求)中进行详述。因而,本发明化合物包括本文详述的任何化合物。本发明包括本文详述的所有化合物的应用,所述化合物包括被描述为组胺受体调节剂的化合物的任何和所有的立体异构体、盐和溶剂化物。使用本发明化合物的进一步的方法的详述贯穿本文。本发明包括式(I)化合物:其中:R1是H、羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R2a和R2b一起形成羰基部分;R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基或硝基或R3a和R3b一起形成羰基部分;X7、X8、X9和X10各自独立地是N或CR4;m和q独立地是0或1;X1是N或CH;R4各自独立地是H、羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基;R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H、羟基、C1-C8烷基或与其所连接的碳及一个孪位的R8(a-f)一起形成环烷基部分或羰基部分;R10a和R10b各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、羟基、烷氧基或R10a和R10b一起形成羰基部分;Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;条件是:(i)当X1是N时,所述化合物不是表1中的化合物或其盐;且(ii)所述化合物不是在U.S.专利6,187,785、7,071,206、3,409,628和6,849,640或在PCT公开WO2005/055951、WO2007/041697和WO2007/087425中所列出的那些化合物。在另一个实施方案中,本发明化合物及使用本文详述的化合物的方法包括式(I)的化合物中的任何一个,其包括表1中所列出的那些化合物或其盐。在一个实施方案中,化合物是其中R4不是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、芳氧基或芳烷基的式(I)化合物。在一个实施方案中,化合物是其中R4不是被取代的或未被取代的芳基的式(I)化合物。在另一个实施方案中,化合物是其中X7、X8、X9和X10中至少一个是N的式(I)化合物,条件是该化合物不是化合物233x、234x或235x或其盐或溶剂化物。在另一个实施方案中,化合物是其中X7、X8、X9和X10中至少一个是N且R1是甲基的式(I)化合物。在一个实施方案中,当化合物具有式(I)的结构且X1是CH,那么所述化合物是式(AA)化合物,其中X7-X10、R2a、R2b、R3a、R3b、R8a-R8f、R10a、R10b、m、q和Q如在式(I)中所定义。在一个实施方案中,化合物是式(I)化合物,其中所述化合物进一步是1型化合物。在另一个实施方案中,化合物是式(I)化合物,其中所述化合物进一步是2型化合物。在另一个实施方案中,化合物是式(I)化合物,其中所述化合物进一步是3型化合物。在又一个实施方案中,化合物是式(I)化合物,其中所述化合物进一步是4型化合物。在一个实施方案中,化合物是式(AA)化合物或其可药用盐:其中:X7-X10、R2a、R2b、R3a、R3b、R8a-R8f、R10a、R10b、m、q和Q如在式(I)的一个实施方案中和在式(E)的另一个实施方案中所定义且:(a)Ra和Rb各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,条件是Ra和Rb基团不都是H,且(i)-(iii)中一项或多项适用:(i)当m和q是0时,Ra和Rb中至少一个不是甲基;(ii)当m和q是0时,X7-X10中至少一个不是CH;(iii)当m和q是0时,(a)Q不是被取代的或未被取代的苯基或(b)Q是被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;或(b)Ra和Rb与它们所连接的氮原子一起形成杂环或被取代的杂环。在式(AA)的一个实施方案中,m和q中至少一个是1且(a)Ra和Rb基团各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,条件是Ra和Rb基团不都是H;或(b)Ra和Rb与氮原子一起形成杂环或被取代的杂环。在另一个实施方案中,化合物是式(AB)的化合物或其可药用盐:其中:X7-X10、R1、R3a、R3b、R8b-R8f、R10a、R10b、m、q和Q如在式(I)的一个实施方案中和在式(E)的另一个实施方案中所定义且:(a)Ra和Rb各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,条件是当m和q是0时,(i)-(v)中的一项或多项适用:(i)R8e和R8f中至少一个是羟基、C1-C8烷基,或R8e和R8f与它们所连接碳一起形成环烷基部分或羰基部分;(ii)Q是被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;(iii)Ra和Rb不是被取代的烷基;(iv)X7-X10中至少一个不是CH;和(v)R1是羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、酰基氨基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;或(b)Ra和Rb与它们所连接的氮原子一起形成杂环或被取代的杂环。在式(AB)的一个实施方案中,Q不是被取代的苯基。在式(AB)的另一个实施方案中,Ra和Rb各自独立地选自H、烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环。在又一个实施方案中,化合物是式(AC)化合物或其可药用盐:其中:X7-X10、R1、R2a、R2b、R3a、R3b、R8a-R8f、m、q和Q如在式(I)的一个实施方案中和在式(E)的另一个实施方案中所定义且:(a)Ra和Rb各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,条件是当m和q是0时,(i)-(v)中一项或多项适用:(i)R8e和R8f独立地是H、羟基、C1-C8烷基或R8e和R8f与它们所连接的碳一起形成环烷基部分;(ii)Q是未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;(iii)Ra和Rb不是被取代的烷基;(iv)X7-X10中至少一个不是CH;和(v)R1是羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、酰基氨基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;或(b)Ra和Rb与它们所连接的氮原子一起形成杂环或被取代的杂环。在式(AC)的一个特定的实施方案中,Q不是未被取代的苯基。在式(AC)的另一个实施方案中,Ra和Rb各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环基和被取代的杂环基。在一个实施方案中,本发明包括式(H)化合物或其盐:其中:R1是H、羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基、羟基、烷氧基、硝基、被取代的或未被取代的氨基,R2a和R2b一起形成羰基部分或R2a和R2b与它们所连接的碳一起形成环烷基部分;R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R3a和R3b一起形成羰基部分或R3a和R3b与它们所连接的碳一起形成环烷基部分;X7、X8、X9和X10各自独立地是N或CR4;m和q独立地是0或1;R4各自独立地是H、羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、羰基烷氧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基;R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H、羟基、C1-C8烷基、C1-C8全卤代烷基、羧基、羰基烷氧基,或与其连接的碳及一个孪位的R8一起形成环烷基部分或羰基部分;R10a和R10b各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、羟基、烷氧基、氰基、硝基、被取代的或未被取代的氨基,R10a和R10b一起形成羰基或R10a和R10b与它们所连接的碳一起形成环烷基部分;Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;条件是正好R1、R2a、R2b、R10a和R10b中的一个是被取代的或未被取代的氨基。在另一个实施方案中,本发明包括式(A)化合物:其中:R1是H、羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R2a和R2b一起形成羰基部分;R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基或硝基或R3a和R3b一起形成羰基部分;X7、X8、X9和X10各自独立地是N或CR4;m和q独立地是0或1;R4各自独立地是H、羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、羰基烷氧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基;R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H、羟基、C1-C8烷基或与其所连接的碳及一个孪位的R8(a-f)一起形成环烷基部分或羰基部分;R10a和R10b各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、羟基、烷氧基或R10a和R10b一起形成羰基;且Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;条件是该化合物不是(i)表1中的化合物或其盐且(ii)该化合物不是化合物229H、230、241、255、256、262和274中任何一个或其盐。在另一个实施方案中,本发明化合物及本文详述的使用所述化合物及施用所述化合物的方法包括任何式A化合物,其包括表1中列出的那些化合物或其盐和化合物229H、230、241、255、256、262和274或其盐。在又一个实施方案中,化合物是式(A)化合物,条件是该化合物不是(i)表1中的化合物或其盐且(ii)该化合物不是化合物229H、230、241、255、256、262和274中任何一个或其盐和(iii)表1A中的化合物或其盐。在一个实施方案中,化合物是式(A)化合物,条件是:(i)它不是表1中的化合物或其盐;且(ii)所述化合物不是在U.S.专利6,187,785、7,071,206、3,409,628和6,849,640或在PCT公开WO2005/055951、WO2007/041697和WO2007/087425中所列出的那些化合物。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式A化合物,其包括表1或表1A中列出的那些化合物或其盐。在一个实施方案中,化合物是式(A)化合物,其中R4不是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、芳氧基或芳烷基。在一个实施方案中,化合物是式(A)化合物,其中R4不是被取代的或未被取代的芳基。在另一个实施方案中,化合物是式(A)化合物,其中X7、X8、X9和X10中至少一个是N,条件是该化合物不是化合物233x、234x或235x或其盐或溶剂化物。在另一个实施方案中,化合物是式(A)化合物,其中X7、X8、X9和X10中至少一个是N且R1是甲基。在另一个实施方案中,本发明包括式(E)化合物:其中:R1是H、羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R2a和R2b一起形成羰基部分或R2a和R2b与它们所连接的碳一起形成环烷基部分;R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R3a和R3b一起形成羰基部分或R3a和R3b与它们所连接的碳一起形成环烷基部分;X7、X8、X9和X10各自独立地是N或CR4;m和q独立地是0或1;R4各自独立地是H、羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、羰基烷氧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基;R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H、羟基、C1-C8烷基、C1-C8全卤代烷基、羧基、羰基烷氧基,或与其所连接的碳及一个孪位的R8一起形成环烷基部分或羰基部分;R10a和R10b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R10a和R10b一起形成羰基部分或R10a和R10b与它们所连接的碳一起形成环烷基部分;且Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的环烷基、被取代的或未被取代的环烯基或被取代的或未被取代的杂环基;条件是(i)该化合物不是表1中的化合物或其盐;(ii)该化合物不是表1A的化合物或其盐且(iii)该化合物不是化合物229H、230、241、255、256、262和274中任何一个或其盐。在另一个实施方案中,本发明化合物及本文详述的使用所述化合物及施用所述化合物的方法包括任何式(E)化合物,其包括表1和表1A中所列的化合物或其盐和化合物229H、230、241、255、256、262和274或其盐。在一个实施方案中,Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-C8环烷基、被取代的或未被取代的C3-C8环烯基或被取代的或未被取代的杂环基。在另一个实施方案中,化合物是式(E)化合物,其中进一步的条件是当Q是被取代的或未被取代的杂芳基时,它不是噻唑、三唑或二唑。然而,在另一个实施方案中,化合物是式(E)化合物且包括其中Q是噻唑、三唑或二唑的化合物。在又一个实施方案中,化合物是式(E)化合物,其中进一步的条件是该化合物不是式(G)化合物。然而,在另一个实施方案中,化合物是式(E)化合物且包括与式(G)一致的化合物。在一个实施方案中,化合物是式(E)化合物,其中所述化合物进一步是1型化合物。在另一个实施方案中,化合物是式(E)化合物,其中所述化合物进一步是2型化合物。在另一个实施方案中,化合物是式(E)化合物,其中所述化合物进一步是3型化合物。在又一个实施方案中,化合物是式(E)化合物,其中所述化合物进一步是4型化合物。在另一个实施方案中,本发明包括式(A)或(E)的化合物,其中m和q中至少一个是1且其中Q是被取代的苯基。在一个实施方案中,Q是被0至6个R9基团取代的苯基,其中R9各自独立地是卤素、氰基、硝基、全卤代烷基、全卤代烷氧基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、酰基、酰氧基、羰基烷氧基、烷硫基、被取代的或未被取代的杂环基、烷氧基、被取代的或未被取代的氨基、酰基氨基、磺酰基氨基、磺酰基、羰基、氨基酰基或氨基羰基氨基。在另一个实施方案中,Q是被至少一个烷基、全卤代烷基、卤素、羟基或烷氧基所取代的苯基。在又一个实施方案中,Q是被至少一个烷基、全卤代烷基、卤素、氰基、硝基、羟基、烷氧基或全卤代烷氧基所取代的苯基。在另一个该类实施方案中,Q是被两个或三个选自甲基、全卤代烷基、卤素、羟基和烷氧基的基团所取代的苯基。在一个特定的实施方案中,m和q中至少一个是1,且Q是被以下基团中至少一个所取代的苯基:甲基、三氟甲基、氟、氯、羟基、甲氧基、乙氧基和异丙氧基。当Q是被两个或三个基团所取代的苯基时,所述基团可以是相同或不同的。例如,当m和q中至少一个是1时,Q在一个实施方案中是二氟苯基、二氯苯基、二甲氧基苯基、二(三氟甲基)苯基、三氟苯基、(氟)(氯)苯基、(氟)(三氟甲基)苯基、(氯)(三氟甲基)苯基或(氟)(甲氧基)苯基。在一个实施方案中,当化合物是式(A)或(E)的化合物且Q是被取代的苯基时,(各)取代基可位于苯环上任何可用的位置。例如,单-取代的苯基可以是在苯基的邻、间或对位被取代。任何可行的苯基取代型式都适用于二-或三-取代的苯基(例如,在邻位和对位、在两个邻位、在两个间位、在间位和对位、在邻、间和对位、在两个邻位和一个对位、在两个邻位和一个间位或在两个间位和一个对位或邻位)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q中的一个是1且另一个是0且Q是被取代的苯基。在另一个实施方案中,化合物式(A)或(E)的化合物,其中m和q都是1且Q是被取代的苯基。在另一个实施方案中,本发明包括式(A)或(E)的化合物,其中m和q是0,Q是被取代的苯基,其中取代基位于苯环的邻位和/或间位,且对位带有一个氢,且R1是甲基或乙基。在另一个实施方案中,本发明包括式(A)或(E)的化合物,其中m和q是0,Q是被取代的苯基,其中取代基位于苯环的邻位和/或间位,且对位带有一个氢,且R1是甲基或乙基,条件是Q不是3-甲基苯基。在一个实施方案中,Q是在邻位或间位单取代的苯基。在一个实施方案中,Q是在邻位单取代的苯基。适合的苯基取代基和取代型式包括上段中详述的那些。在另一个实施方案中,本发明包括式(A)或(E)的化合物,其中m和q是0,Q是被取代的苯基且R1是甲基,条件是(i)-(iii)中至少一项适用:(i)当R8e和R8f与它们所连接的碳一起形成羰基时,X9不是CR4,其中R4是甲基;(ii)Q是具有至少一个氟取代基的被取代的苯基;和(iii)当Q是4-甲氧基苯基时,X9不是CR4,其中R4是氯。在另一个实施方案中,本发明包括式(A)或(E)的化合物,其中m和q是0,Q是被取代的苯基且R1是甲基,条件是(i)-(iii)中至少一项适用:(i)当R8e和R8f与它们所连接的碳一起形成羰基时,X9不是CR4,其中R4是OCH3;(ii)Q是具有至少一个氟取代基的被取代的苯基,条件是Q不是4-氟苯基;和(iii)当Q是4-甲氧基苯基时,X9不是CR4,其中R4是氯。在一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q中的一个是1且另一个是0,且R8e和R8f与它们所连接的碳一起形成羰基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q中的一个是1且另一个是0,且R8e和R8f与它们所连接的碳一起形成羰基,条件是(i)R1不是异丙基和/或(ii)Q不是被取代的哌嗪基。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q中的一个是1且另一个是0,R8e和R8f与它们所连接的碳一起形成羰基,且R1是CH3或H。在又一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q中的一个是1且另一个是0,R8e和R8f与它们所连接的碳一起形成羰基,R1是CH3或H,X7、X8和X10是CH且X9是CR4,其中R4是卤素(例如,氯、溴、碘)或烷基(例如,甲基)。在又一个此类实施方案中,R1是CH3或H,X7、X8和X10是CH,X9是CR4,其中R4是卤素(例如,氯、溴、碘)或烷基(例如,甲基)且R2a、R2b、R3a、R3b、R10a和R10b各自是氢。在又一个此类实施方案中,其中Q是被取代的或未被取代的哌嗪基,R1不是异-丙基。在又一个此类实施方案中,其中Q是被取代的或未被取代的哌嗪基,R1是H、CH3或乙基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q都是1且:(a)X7-X10中至少一个不是CH或(b)Q是选自被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基和被取代的或未被取代的杂环基的单环。在一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q都是1且:(a)X7-X10中至少一个不是CH和OCH3或(b)Q是选自被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基和被取代的或未被取代的C3-8环烯基的单环。在一个特定的实施方案中,化合物是式(A)或(E)的化合物,其中m和q都是1且X7-X10中至少一个是CR4,其中R4是卤素或烷基。在又一个实施方案中,m和q都是1;X7-X10中至少一个是CR4,其中R4是卤素或烷基;R1是烷基且R2a、R2b、R3a、R3b、R10a和R10b各自是氢。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R8a-R8f中至少一个,在当其存在时,是OH。由此,当m是0且q是1,R8a、R8b、R8e和R8f中至少一个是OH。在又一个实施方案中,化合物是式(A)或(E)的化合物,其中R8a-R8f中的一个是OH且该基团孪位的R8(a-f)基团是烷基或全卤代烷基。在另一个此类实施方案中,m和q中的一个是1且另一个是0;R8a-R8f中的一个是OH且该基团孪位的R8(a-f)基团是烷基(例如,甲基、乙基、环丙基)或全卤代烷基(例如,三氟烷基);X7、X8和X10各自是CH;X9是CR4,其中R4是烷基(例如,甲基)或卤素(例如,氯、氟);且R1是烷基(例如,甲基、环丙基)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q都是0且:(a)Q是苯基且R1是乙基;(b)Q是6-甲基-3-吡啶基且R8e和R8f都是H;或(c)Q是6-甲基-3-吡啶基且X9是C-R4,其中R4是羟基、硝基、氰基、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、羰基烷氧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q都是0,Q是6-甲基-3-吡啶基且X9是C-R4,其中R4是氢或烷基(例如,甲基)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中X7-X10中至少一个是N,且(i)-(iii)中至少一项适用:(i)R8e和R8f都是H;(ii)R1是甲基;和(iii)Q是被取代的或未被取代的吡啶基。在一个此类实施方案中,当Q是被取代的吡啶基时,其是被烷基(例如,甲基、乙基、异丙基、正丙基、环丙基)、全卤代烷基(例如,三氟甲基)、被取代的烷基(例如,羟基甲基)、羧基、羰基烷氧基(例如,CO2CH3、CO2CH2CH3)、被取代的氨基(例如,NHCH3、N(CH3)2、NHCH(CH3)2)、氨基酰基(例如,NHC(O)CH3)、烷氧基(例如,OCH3、OCH2CH3)、N-氧化物(例如,当吡啶基的环氮被氧化)、稠合于该吡啶基的芳基或杂芳基、卤素(例如,F、Cl、Br)、羟基、N-烷基(例如,当吡啶基的N被甲基或环丙基所取代)、N-全卤代烷基(例如,其中吡啶基的N被三氟甲基所取代)、氧代(当位于吡啶环的环氮的邻位时,其可作为互变异构体存在)所取代。所述被取代的吡啶基优选地是被一个或两个取代基所取代。取代基可位于吡啶基环的任何可用的位置。例如,单取代的3-吡啶基的取代基可位于2、4、5或6位。在一个实施方案中,化合物是式(A)或(E)的化合物,其中X9是C-R4,其中R4是-COOH,条件是当R1是甲基时,Q不是6-甲基-3-吡啶基或4-吡啶基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q中的一个是1且另一个是0,X9是C-R4,其中R4是-COOH,条件是当R1是甲基时,Q不是6-甲基-3-吡啶基或4-吡啶基。在另一个实施方案中,当X9是C-R4且R1是H时,Q不是6-甲基-3-吡啶基。然而,在又一个实施方案中,本发明的方法、药物组合物、药盒和纯化或分离形式的化合物包括式(A)或(E)的化合物,当X9是C-R4时,R1是H且Q是6-甲基-3-吡啶基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是三氟甲基吡啶基。在又一个实施方案中,m和q中的一个是1且另一个是0,Q是三氟甲基-取代的吡啶基(例如,4-三氟甲基-3-吡啶基、5-三氟甲基-3-吡啶基、6-三氟甲基-3-吡啶基)且R1是甲基。在又一个实施方案中,m和q中的一个是1且另一个是0,Q是三氟甲基-取代的吡啶基(例如,4-三氟甲基-3-吡啶基、5-三氟甲基-3-吡啶基、6-三氟甲基-3-吡啶基),R1是甲基且X7-X10中至少一个不是CH(例如,当X7-X10中的一个是CR4,其中R4是烷基(诸如甲基、乙基、异丙基和叔-丁基)、卤素(诸如氯和氟)、羧基(COOH)或全卤代烷氧基(诸如-OCF3)。在又一个实施方案中,m和q中的一个是1且另一个是0,R1是甲基,X9是CR4,其中R4是卤素或CH3,且Q是被取代的或未被取代的6-三氟甲基-3-吡啶基。在又一个实施方案中,m和q中的一个是1且另一个是0,R1是甲基且X9是CR4,其中R4是卤素或CH3,且Q是被取代的6-三氟甲基-3-吡啶基。在又一个实施方案中,m和q中的一个是1且另一个是0,R1是甲基且X9是CR4,其中R4是卤素或CH3且Q是6-三氟甲基-3-吡啶基。在另一个此类实施方案中,Q是被一个、两个或三选自以下的基团所取代的6-三氟甲基-3-吡啶基:卤素、氰基、硝基、全卤代烷基、全卤代烷氧基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、酰基、酰氧基、羰基烷氧基、烷硫基、被取代的或未被取代的杂环基、烷氧基、被取代的或未被取代的氨基、酰基氨基、磺酰基氨基、磺酰基、羰基、氨基酰基和氨基羰基氨基。在另一个此类实施方案中,Q是被一个、两个或三选自以下的基团所取代的6-三氟甲基-3-吡啶基:卤素、氰基、硝基、全卤代烷基和全卤代烷氧基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R1是环丙基、被取代的苯基或羰基烷氧基,条件是当R1是羰基烷氧基时,m和q中至少一个是1。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R1是环丙基,X7、X8和X10各自是CH且X9是CR4,其中R4是烷基或卤素。在另一个此类实施方案中,化合物是式(A)或(E)的化合物,其中R1是环丙基,X7、X8和X10各自是CH;X9是CR4,其中R4是烷基或卤素,且m和q中的一个是1且另一个是0。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中R1是被取代的苯基且Q是6-甲基-3-吡啶基。在又一个实施方案中,化合物是式(A)或(E)的化合物,其中R1是被取代的苯基,Q是6-甲基-3-吡啶基,X7、X8和X10各自是CH;且X9是CR4,其中R4是烷基(例如,甲基)或卤素(例如,氯)。在该实施方案的一个方面,m和q中的一个是1且另一个是0。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R2a与R2b或者R10a与R10b与它们所连接的碳一起形成羰基且m和q中至少一个是1。在一个特定的此类实施方案中,R2a和R2b与它们所连接的碳一起形成羰基,X7、X8和X10各自是CH;且X9是CR4,其中R4是烷基(例如,甲基),且Q是未被取代的苯基或被取代的吡啶基(例如,6-甲基-3-吡啶基和6-CO2H-3-吡啶基)。在一个实施方案中,当R2a和R2b与它们所连接的碳一起形成羰基时,X7、X8和X10各自是CH,X9是CR4,其中R4是烷基(例如,甲基),且Q不是6-CO2H-3-吡啶基。然而,本发明的方法、药物组合物、药盒和纯化或分离形式的化合物包括该实施方案的化合物,其包括其中R2a和R2b与它们所连接的碳一起形成羰基,X7、X8和X10各自是CH,X9是CR4,其中R4是烷基(例如,甲基),且Q是6-CO2H-3-吡啶基的那些化合物。在另一个此类实施方案中,R10a和R10b与它们所连接的碳一起形成羰基;X7、X8和X10各自是CH或N且X9是N或CR4,其中R4是烷基(例如,甲基)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中X9是C-R4,其中R4是三氟甲基和Q是被取代的杂芳基。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中X9是C-R4,其中R4是三氟甲基,X7、X8和X10是CH或N;且Q是被取代的或未被取代的芳基、被取代的杂芳基或未被取代的杂环基部分。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中X9是C-R4,其中R4是三氟甲基,且Q是被取代的杂芳基。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中X9是C-R4,其中R4是三氟甲基,X7、X8和X10是CH或N;m和q中的一个是1且另一个是0,且Q是被取代的芳基、被取代的杂芳基或未被取代的杂环基部分。在一个实施方案中,化合物是式(A)或(E)的化合物,其中X9是C-R4,其中R4是异丙基或Q是被取代的或未被取代的吡嗪基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中X9是C-R4,其中R4是异丙基或Q是被取代的或未被取代的吡嗪基,条件是当Q是2-吡嗪基时,(i)R9不是CR4,其中R4是CH3或(ii)R2a和R2b中至少一个不是氢。在一个实施方案中,X9是C-R4,其中R4是异丙基;X7、X8和X10是CH,且R1是甲基。在另一个此类实施方案中,Q是被取代的或未被取代的吡嗪基;X7、X8和X10是CH;X9是C-R4,其中R4是卤素(例如,氯)或烷基(例如,甲基、乙基、异丙基),且R1是甲基。在又一个此类实施方案中,Q是被取代的或未被取代的吡嗪基;X7、X8和X10是CH;X9是C-R4,其中R4是卤素(例如,氯)或C2-C4烷基(例如,乙基、异丙基),且R1是甲基。在另一个此类实施方案中,m和q中的一个是1且另一个是0。在一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q中的一个是1且另一个是0,R8e和R8f都是H且:(i)Q是被取代的哌嗪基,其中所述被取代的哌嗪基不是4-(2-甲氧基苯基)-哌嗪基或(ii)Q是被取代的哌嗪基且X7-X10中至少一个不是CH。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中m和q中的一个是1且另一个是0,R8e和R8f都是H且:(i)Q是被取代的哌嗪基,其中所述被取代的哌嗪基不是4-(2-甲氧基苯基)-哌嗪基或(ii)Q是被取代的哌嗪基且X7-X10中至少一个不是CH或CR4,其中R4是OCH3。在一个此类实施方案中,m和q中的一个是1且另一个是0;R8e和R8e都是H;Q是被取代的哌嗪基;X7、X8和X10是N或CR4,其中R4是H或F,且X9是CR4,其中R4是烷基(例如,甲基)或卤素(例如,氯)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是6-甲基-3-吡啶基;m和q中的一个是1且另一个是0;R1是甲基;且X9是CR4,其中R4是碘或被取代的烷基。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是6-甲基-3-吡啶基;m和q中的一个是1且另一个是0;R1是甲基;且R8e和R8f中至少一个是羟基或烷基,或R8e和R8f与它们所连接的碳一起形成环烷基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是羧基取代的吡啶基,条件是其不是6-羧基-3-吡啶基,或是羰基烷氧基取代的吡啶基。然而,在又一个实施方案中,本发明的方法、药物组合物和纯化的或分离形式的化合物包括式(A)或(E)化合物,其中Q是6-羧基-3-吡啶基。在特定的实施方案中,化合物进一步具有一个或多个以下的结构特征:R1是C2-C6烷基,和X9不是CR4,其中R4是甲基。在一个实施方案中,R1是C2-C6烷基,X9不是CR4,其中R4是甲基,且Q是羧基取代的吡啶基或羰基烷氧基取代的吡啶基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是被取代的苯基且m和q中至少一个是1。在一个特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:R1是甲基或乙基;X7、X8和X10是CH;和X9是CR4,其中R4是卤素、烷基、H或全卤代烷基(例如,Cl、I、F、CF3、甲基)。在又一个实施方案中,Q是被取代的苯基;m和q中至少一个是1且R1是甲基。在又一个实施方案中,Q是被取代的苯基;m和q中至少一个是1;R1是甲基且(i)-(iii)中的一项或多项适用:(i)X7、X8和X10各自独立地是CR4,其中R4是氢或卤素(例如,氯);(ii)X9是CR4,其中R4是氢、烷基、卤素、全卤代烷基或全卤代烷氧基;和(iii)R2a和R2b独立地是氢、甲基或氟。适合的苯基取代基和取代型式包括但不限于本文详述的那些,例如,第66页第三段所述。在一个特定的实施方案中,苯基取代基是定义为R9的那些。在式(E)的一个特定的实施方案中,m和q中的一个是1且另一个是0;R1是CH3;X7是N或CR4,其中R4是氢或氯;X8和X10独立地是CR4,其中R4是氢或氯;X9是CR4,其中R4是烷基、卤素、全卤代烷基或全卤代烷氧基,且Q是被取代的或未被取代的芳基、被取代的或未被取代的环烷基或被取代的或未被取代的环烯基。在另一个此类实施方案中,R2a、R2b、R3a、R3b、R10a和R10b各自是氢。在一个实施方案中,当Q是苯基,那么X7、X8和X10中至少一个是氯。在另一个实施方案中,当Q是苯基,那么R2a、R2b、R3a、R3b、R10a、R10b和R8a-R8f中至少一个不是氢。在又一个实施方案中,当Q是苯基,那么X9是CR4,其中R4是氯、溴或碘。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是未被取代的苯基且m和q中至少一个是1,条件是当m和q中只有一个是1,X9是CR4且X7、X8和X10是CH,那么R4不是H。在一个此类实施方案中,当Q是未被取代的苯基且m和q中只有一个是1,那么X9不是CH。在特定的实施方案中,化合物进一步包含R1,其是烷基(例如,甲基、乙基、环丙基)或H。在一些此类实施方案中,X9是C-R4,其中R4是氢、烷基(例如,甲基)或卤素(例如,氯)。在其他的此类实施方案中,X7-X10中至少一个是C-R4,其中R4是卤素(例如,氯)。在这些实施方案的另一些中,X7-X10中只有一个是C-R4,其中R4是卤素,且其他的各自是CH。在另一个此类实施方案中,当X9是C-R4,其中R4是氢,那么X7、X8或X10中至少一个是卤素(例如,氯)。在又一个实施方案中,Q是未被取代的苯基,m和q中至少一个是1且X7-X10中至少一个是N。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R8a-R8f中任何一个,当其存在时,是羟基。在一个实施方案中,R8e和R8f中的一个是OH。在另一个实施方案中,当R8e和R8f中的一个是OH,另一个是甲基。在又一个实施方案中,R8a-R8f中至少一个,当其存在时,是羟基,且m和q中的一个是1且另一个是0。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:R1是甲基;Q是被取代的或未被取代的吡啶(例如,3-吡啶基、4-吡啶基和6-甲基-3-吡啶基);和X9是CR4,其中R4是卤素(例如,氯)或烷基(例如,甲基)。在一个特定的实施方案中,R8e和R8f中的一个是OH;X7、X8和X10各自是C-R4,其中R4是氢;且X9是C-R4,其中R4是烷基(例如,甲基)或卤素(例如,氯和氟)。在另一个实施方案中,m是1,q是0,且R8c-R8f中的任何一个是羟基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R8e和R8f与它们所连接的碳一起形成羰基且:(i)Q是(a)被取代的或未被取代的哌啶基(其在一个实施方案中经它的环氮原子连接于母体结构)或(b)未被取代的哌嗪基或被支链的烷基(例如,异丙基)取代的哌嗪基,或(ii)X9是CR4,其中R4是卤素(例如,氯)。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:R1是甲基,以及m和q中至少一个是1。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中R8e和R8f与它们所连接的碳一起形成羰基;R1是甲基或氢且m和q中的一个是1且另一个是0。在另一个此类实施方案中,化合物进一步具有一种或多种以下的结构特征:X7、X8和X10各自是CH;X9是CR4,其中R4是卤素或烷基(例如,甲基);和R2a、R2b、R3a、R3b、R10a和R10b各自是H。在一个实施方案中,化合物是式(A)或(E)的化合物,其中X7-X10中至少一个是CR4,其中R4是卤素,且Q是哌啶基。在一个此类实施方案中,化合物进一步具有以下的结构特征:X9是CR4,其中R4是卤素(例如,氯),和Q是1-哌啶基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是被取代的或未被取代的哌啶基且X7-X10中至少一个是N或CR4,其中R4是卤素。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是被取代的或未被取代的哌啶基且R8e和R8f与它们所连接的碳一起形成羰基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R8e和R8f与它们所连接的碳一起形成羰基,m是1且q是0;且Q是被取代的或未被取代的杂芳基或未被取代的杂环基;条件是当Q是未被取代的哌嗪基时,R1不是异-丙基(例如,H、CH3或乙基)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R8e和R8f与它们所连接的碳一起形成羰基,m是1且q是0;且Q是被取代的或未被取代的杂芳基或被取代的或未被取代的杂环基;条件是当Q是被取代的或未被取代的哌嗪基时,R1不是异-丙基(例如,H、CH3或乙基)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中R8e和R8f与它们所连接的碳一起形成羰基,m是1且q是0;R1是氢或甲基且Q是未被取代的杂环。在一个此类实施方案中,未被取代的杂环包含不超过一个环杂原子。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中R8e和R8f与它们所连接的碳一起形成羰基;R1是甲基;Q是被取代的或未被取代的杂芳基或未被取代的杂环基且m和q中的一个是1且另一个是0。在另一个此类实施方案中,化合物进一步具有一种或多种以下的结构特征:X9是CR4,其中R4是卤素或烷基(例如,甲基)。在另一个此类实施方案中,化合物进一步具有一种或多种以下的结构特征:X7、X8和X10各自是CH;X9是CR4,其中R4是卤素或烷基(例如,甲基);和R2a、R2b、R3a、R3b、R10a和R10b各自是H。在一个此类实施方案中,Q是在另一个实施方案中,化合物是式(B-1)化合物:其中:R4是卤素或CH3;Q是被取代的或未被取代的杂芳基或被取代的或未被取代的杂环基;且X7、X8、X10、R8c和R8d如在式(B)的一个实施方案中和式(E)的另一个实施方案中所定义。在B-1的一个实施方案中,Q是:在另一个实施方案中,本发明包括式(B-2)化合物其中:R4是卤素或CH3;且R9各自独立地是氢、卤素、氰基、硝基、全卤代烷基或全卤代烷氧基。在另一个此类实施方案中,R4是卤素或CH3且Q是3Py-6CF3。在又一个实施方案中,化合物是式(B)或(E)的化合物,条件是当化合物是式(E)化合物时,m是1且q是0,且条件是(i)-(iv)中至少一项适用:(i)当X7-X10各自是CR4,其中R4是氢,且R1是甲基或叔-丁基,那么Q不是苯基或未被取代的吡啶基;(ii)当X7、X8和X10各自是CR4,其中R4是氢,且X9是CR4,其中R4是甲基,且R1是甲基,那么Q不是苯基、未被取代的吡啶基或吡嗪基;(iii)当X7、X8和X10各自是CR4,其中R4是氢,且X9是CR4,其中R4是氟或三氟,且R1是甲基,那么Q不是苯基、未被取代的吡啶基和6-甲基-3-吡啶基;和(iv)当X8、X9和X10各自是CR4,其中R4是氢,且X7是CR4,其中R4是氟或三氟,且R1是甲基,那么Q不是苯基或3-吡啶基。在另一个实施方案中,化合物是式(B)或(E)化合物,条件是当化合物是式(E)化合物时,m是1和q是0,且条件是(i)和(ii)中至少一项适用:(i)当X8和X10都是CH,X7和X9中的一个是CR4,其中R4是F或CF3,且X7和X9中的另一个是CH,且R1是被取代的C1-C3烷基,其中取代基是羧基或羰基烷氧基,那么Q不是未被取代的苯基、4-氟苯基和未被取代的吡啶基;或(ii)当X7-X10各自是CH且R1是羰基烷氧基,那么Q不是未被取代的苯基和未被取代的吡啶基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中X7-X10中至少一个不是CH且Q是吡咯烷基。在一个实施方案中,X7-X10中至少一个是C-R4,其中R4是烷基,且Q是吡咯烷基。在一个特定的实施方案中,X9是CR4,其中R4是甲基,Q是吡咯烷基,且X9是CR4,其中R4是烷基(例如,甲基)、全卤代烷基(例如,CF3)或卤素(例如,氯)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是吗啉代,且(i)-(iii)中至少一项适用:(i)m和q中至少一个是0;(ii)X7-X10中至少一个不是CH;和(iii)X7-X10中至少一个是CR4,其中R4是卤素。在一个特定的实施方案中,Q是吗啉代,X9是CR4,其中R4是氯,且m和q中至少一个是1且另一个是0。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是吗啉代,R1是烷基且以上(i)-(iii)中至少一项适用。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是全卤代烷基取代的吡啶基(例如,其中Q是在5或6位被CF3取代的3-吡啶基)。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:X7-X10中至少一个是CR4,其中R4各自独立地是烷基(例如,C1-C3烷基,诸如甲基、乙基和异丙基)或卤素(例如,氯或氟);X7、X8或X9中至少一个是CR4,其中R4各自独立地是烷基(例如,C1-C3烷基,诸如甲基、乙基和异丙基)或卤素(例如,氯或氟);X7-X10中的两个是CR4,其中R4各自独立地是烷基(例如,C1-C3烷基,诸如甲基、乙基和异丙基)或卤素(例如,氯或氟),X7-X10中的两个是CR4,其中R4各自独立地是卤素(例如,氯或氟);X8和X9都是CR4,其中R4各自独立地是卤素(例如,氯或氟),所述卤素取代基可以是相同的或不同的(例如,X8是C-F且X9是C-Cl或X8是C-F且X9是C-Cl);以及R1是甲基,和m和q中至少一个是1。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是被取代的或未被取代的具有至少两个环N原子的6-元杂芳基。在一个实施方案中,Q是5-嘧啶基或2-吡嗪基。在另一个实施方案中,当Q是吡嗪基,X9是CR4,其中R4是卤素或C2-C4烷基。在一个实施方案中,当Q是被取代的6-元杂芳基,其是被一个烷基取代,例如,甲基(例如,2-甲基-5-嘧啶基或5-甲基-2-吡嗪基)。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:R1是甲基;X9是CR4,其中R4是卤素(例如,氯)或烷基(例如,甲基、乙基、异丙基);以及其中m和q中的一个是1且另一个是0。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是环己基。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:m和q中的一个是1且另一个是0;X7-X10中至少一个是CR4,其中R4是卤素(例如,其中X8、X9或X10是CR4,其中R4是卤素;例如,其中X7-X10中的一个是CR4,其中R4是卤素,且其他的是CH);R1是甲基;X7-X10中的一个是CR4,其中R4是烷基(例如,当R4是甲基时;例如,当X9是C-CH3且X7、X8和X10是CH时)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是被取代的或未被取代的吡啶基且R1是异丙基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是被取代的吡啶基,条件是(i)吡啶基取代基不是甲基和(ii)当m和q中的一个是1且另一个是0,X9是CR4,其中R4是甲基,且R1是CH3时,吡啶基取代基不是COOH。然而,在又一个实施方案中,本发明的方法、药物组合物和纯化的或分离形式的化合物包括式(A)或(E)化合物,当Q是被取代的吡啶基且(i)取代基是甲基和(ii)当m和q中的一个是1且另一个是0,X9是CR4,其中R4是甲基,且R1是CH3时,取代基是COOH。在一个特定的实施方案中,Q是被至少一个选自乙基、丙基(正-丙基或异丙基)、卤素(例如,氯)和羟基甲基的基团取代的吡啶基。在另一个特定的实施方案中,Q是被至少一个选自乙基、丙基(正-丙基或异丙基)、卤素(例如,氯)、全卤代烷基(例如,CF3)和羟基甲基的基团取代的吡啶基。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:R1是甲基和X9是CR4,其中R4是甲基或氯(且在一个特定的实施方案中,X7、X8和X10是CH);以及m和q是1且另一个是0。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是甲基吡啶基(例如,6-甲基-3-吡啶基);m和q是0且:(i)X7-X9中至少一个是CR4,其中R4是烷基(例如,甲基)或(ii)R8e和R8e都是H。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:X9是CR4,其中R4是甲基,X7、X8和X10是CH,以及R1是甲基或乙基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中X8和X10中至少一个是CR4,其中R4是F。在一个特定的实施方案中,X8是CR4,其中R4是F;X9是CR4,其中R4是卤素(例如,氯)或氢,且R1是烷基(例如,甲基)。在另一个实施方案中,X10是CR4,其中R4是F,且m和q中的一个是1且另一个是0。在又一个实施方案中,X8和X10中至少一个是CR4,其中R4是F;m和q中的一个是1且另一个是0,且Q是6-三氟甲基-3-吡啶基或6-甲基-3-吡啶基。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是6-甲基-3-吡啶基;R1是甲基,且X7-X10中的一个是C-F。在一个特定的实施方案中,Q是6-甲基-3-吡啶基;R1是甲基;X8是C-F且X9是CR4,其中R4是卤素(例如,氯)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是5-甲基-3-吡啶基。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:m和q中的一个是1且另一个是0;R1是甲基且X9是CR4,其中R4是烷基(例如,甲基)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是未被取代的吡啶基(例如,2-吡啶基)且m和q都是1。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:R1是甲基;X9是CR4,其中R4是烷基(例如,甲基)。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是未被取代的3-吡啶基;m和q中的一个是1且另一个是0,且X9是CR4,其中R4是烷基(例如,甲基)。在一个实施方案中,R1是甲基或羰基烷氧基,条件是当R1是甲基,那么R2a、R2b、R3a、R3a、R8a-R8f和R10a、R10b中至少一个不是氢。在特定的实施方案中,化合物进一步具有一种或多种以下的结构特征:R1是甲基;R2a、R2b、R3a、R3a、R8a-R8f和R10a、R10b各自是H。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是未被取代的2-吡啶基;m和q中的一个是1且另一个是0且:(i)R1是乙基或(ii)X7-X9中至少一个是CR4,其中R4不是H(在一个特定的实施方案中,X9是CR4,其中R4是烷基(例如,甲基)或卤素(例如,氯))。在特定的实施方案中,化合物进一步具有以下的结构特征之一:R1是乙基和X9是CR4,其中R4是CH3,或R1是乙基和X7-X9各自是CR4。在一个实施方案中,化合物是式(A)或(E)的化合物,其中Q是4-吡啶基;m和q中的一个是1且另一个是0;且R1是乙基。在特定的实施方案中,化合物进一步具有以下的结构特征之一:X9是CR4,其中R4是H或烷基以及X7、X8和X10是H。在又一个实施方案中,化合物是式(A)或(E)的化合物,其中X7-X10中至少一个是N且(i)R1是甲基或(ii)R8e和R8f都是氢。在一个此类实施方案中,X7-X10中只有一个是N,R1是甲基,且X7-X10中不是N的基团是CR4,其中R4是氢、甲基或三氟甲基。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中X7-X10中的两个N(例如,X7和X10都是N或X7和X8都是N)。在又一个实施方案中,化合物是式(A)或(E)的化合物,其中q是0,m是1,R8e和R8f一起形成羰基,且(i)-(vi)中至少一项适用:(i)Q是被取代的或未被取代的杂芳基或未被取代的杂环基,(ii)R1是CH3或H,(iii)X9是卤素或CH3,条件是当X9是CH3时,R1是H或CH3,(iv)X7和X8都是CR4(例如,CH)且R1是H或CH3,(v)X7-X10各自是CH且R1是H或CH3,和(vi)X7-X10各自是CH且Q是被取代的或未被取代的杂芳基或未被取代的杂环基。在另一个实施方案中,化合物是式(A)或(E)的化合物,其中q是0,m是1,R8e和R8f一起形成羰基,且Q是未被取代的或被取代的哌啶基。在一个特别的此类实施方案中,Q是未被取代的哌啶基,X9是卤素或CH3且R1是CH3或H。本发明还包括式(B)的化合物:其中:R1是H、羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R2a和R2b一起形成羰基部分;R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基或硝基或R3a和R3b一起形成羰基部分;X7、X8、X9和X10各自独立地是N或CR4;R4各自独立地是H、羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、羰基烷氧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基;R8c、R8d、R8e和R8f各自独立地是H、羟基、C1-C8烷基或与其所连接的碳及一个孪位的R8一起形成环烷基部分或羰基部分;R10a和R10b各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、羟基、烷氧基或R10a和R10b一起形成羰基;且Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;条件是(i)该化合物不是化合物72x、125x、126x、127x、128x、129x、130x、131x、132x、133x、134x、135x、136x、137x、138x、139x、140x、141x、142x、143x、144x、145x、146x、148x、149x、150x、151x、152x、155x、156x、157x、158x、159x、160x、161x、162x、163x、164x、165x、166x、167x、168x、169x、170x、171x、172x、173x、174x、175x、176x、177x、178x、179x、180x、181x、182x、183x、184x、185x、186x、187x、188x、189x、190x、191x、192x、193x、194x、195x、196x、197x、198x、199x、200x、201x、203x、204x、222x、223x、224x、225x、226x、227x、228x、229x、233x、234x、235x、237x、241x、243x、244x、245x、246x、247x、248x、249x、250x、251x、252x、253x、254x、255x、256x、257x和258x中的任何一个或其盐且(ii)该化合物不是化合物229H、230、241、255、256、262和274中的任何一个或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括式B化合物中的任何一个,其包括表1中列出的那些化合物或其盐和化合物229H、230、241、255、256、262和274或其盐。在一个实施方案中,化合物是式(B)化合物,其中所述化合物进一步是1型化合物。在另一个实施方案中,化合物是式(B)化合物,其中所述化合物进一步是2型化合物。在另一个实施方案中,化合物是式(B)化合物,其中所述化合物进一步是3型化合物。在又一个实施方案中,化合物是式(B)化合物,其中所述化合物进一步是4型化合物。在一个实施方案中,化合物是式(B)化合物,条件是(i)Q不是未被取代的吡啶基或4-取代的-1-哌嗪基;(ii)当Q是被取代的吡啶基时,其是被至少一个不是甲基的基团所取代;且(iii)该化合物不是化合物176x、177x、178x、179x、204x、243x、244x、245x、246x、247x和251x中的任何一个。在又一个实施方案中,化合物是式(B)的化合物,条件是(i)Q不是未被取代的吡啶基或4-取代的-1-哌嗪基;(ii)当Q是被取代的吡啶基时,其是被至少一个不是甲基的基团所取代;(iii)当Q是吡咯烷基、哌啶基、氮杂基、苯基或被取代的哌嗪基时,X7、X8、X9和X10中至少一个不是CH;且(iv)该化合物不是化合物176x、178x、243x、244x、245x、246x、247x和251x中任何一个。在又一个实施方案中,化合物是式(B)的化合物,条件是(i)R8c、R8d、R8e和R8f中至少一个不是H;且(ii)R8e和R8f不与它们所连接的碳一起形成羰基。在另一个实施方案中,化合物是式B化合物,其中Q是被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基,条件是:(i)当Q是被取代的哌嗪-1-基时,其不是4-(2-甲氧基苯基)哌嗪-1-基;(ii)当Q是吡咯烷-1-基、哌啶-1-基或氮杂环庚烷-1-基时,其是被取代的吡咯烷-1-基、哌啶-1-基或氮杂环庚烷-1-基或(iii)化合物不是2-甲基-5-(2-(吡咯烷-1-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;2-甲基-5-(2-(哌啶-1-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;2,8-二甲基-5-(2-(哌啶-1-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;或5-(2-(氮杂环庚烷-1-基)乙基)-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚或其盐或溶剂化物。在一个实施方案中,化合物是式B化合物或本文详述的其任何的实施方案,其中Q是碳环,诸如5、6或7元的碳环。在一个实施方案中,化合物是式B化合物或本文详述的其任何的实施方案,其中Q是杂环,诸如3、4、5、6或7元的碳环。在一个实施方案中,化合物是式B化合物或本文详述的其任何的实施方案,其中Q是杂环,诸如5、6或7元的杂环。在一个实施方案中,化合物是式B化合物或本文详述的其任何的实施方案,其中Q是杂环,诸如3、4、5、6或7元的杂环。在另一个实施方案中,化合物是式B化合物,其中Q是被取代的或未被取代的芳基或被取代的或未被取代的杂芳基,条件是(i)X7-X10中至少一个不是CH且(ii)-(viii)中至少一项适用:(ii)当Q是2-甲基吡啶-3-基或选自苯基和吡啶-3-基的未被取代的基团时,R1不是甲基,且条件是该化合物不是125x、177x、184x和237x(iii)当Q是未被取代的吡啶-2-基时,R1不是H或选自甲基、丙基和丁基的未被取代的基团,且条件是该化合物不是180x-183x;(iv)当Q是未被取代的吡啶-4-基时,R1不是H或选自甲基、丙基、丁基、苯乙基和苯甲酰基的未被取代的基团,条件是该化合物不是185x-241x;(v)当Q是6-甲基吡啶-3-基时,R1不是H或选自甲基、乙基、丙基、丁基、异丁基、仲-丁基、戊基、异戊基、环己基、庚基、苄基和苯基的未被取代的基团;(vi)X7-X10中至少一个是N;(vii)R8c-R8f中至少一个不是H;和(viii)R2a、R2b、R3a、R3b、R10a或R10b中至少一个不是H,或其盐或溶剂化物。在另一个实施方案中,化合物是式B化合物,其中Q是被取代的或未被取代的杂芳基,诸如5、6或7元的杂芳基,条件是以上(ii)-(viii)中至少一项适用。在一个实施方案中,化合物是式B化物,其中Q是被取代的或未被取代的芳基,诸如5、6或7元的芳基,条件是:(a)当Q是未被取代的苯基时,R1不是甲基,条件是化合物不是177x;(b)X7-X10中至少一个是N;(c)R8c-R8f中至少一个不是H;或(d)R2a、R2b、R3a、R3b、R10a或R10b中至少一个不是H,或其盐或溶剂化物。在另一个实施方案中,本发明包括式(B)化物,其中X7-X10、R1、R2a、R2b、R3a、R3b、R8c-R8f、R10a、R10b、m和q如在式(I)中的一个实施方案中和在式(E)的另一个实施方案中所定义,且Q是被取代的苯基或被取代的或未被取代的吡咯基,或其可药用盐。在式(B)的一个实施方案中。X9是C-R4,其中R4是卤素或H。在式(B)的另一个实施方案中、R1是烷基,诸如C1-C8烷基,例如,甲基。本发明包括式(B)化合物,其中X9是C-R4,其其中R4是卤素(例如,氯)或H且,其中R1是甲基。在式(B)的另一个实施方案中,X7、X8和X10各自是CH;X9是C-R4,其中R4是卤素或H,且R2a、R2b、R3a、R3b、R8a-R8f、R10a、R10b独立地是H或烷基,例如,甲基。在一个实施方案中,本发明包括式(B)化合物,其中X9是C-R4,其中R4是氯;X7是N或C-R4,其中R4是氢或卤素;X8和X10独立地是C-R4,其中R4是氢或氟,且(i)-(iii)中至少一项适用:(i)R8e和R8f中至少一个是羟基、C1-C8烷基、C1-C8全卤代烷基或与其所连接碳及一个孪位的R8(e-f)一起形成环烷基或羰基;(ii)R8e和R8f中至少一个是C1-C8烷基;(iii)Q是被取代的或未被取代的芳基、被取代的或未被取代的C3-C8环烷基、被取代的或未被取代的C3-C8环烯基或被取代的或未被取代的杂环基。在一个特定的实施方案中,本发明包括式(B)化合物,其中X9是C-R4,其中R4是氯;X7是N或C-R4,其中R4是氢或卤素;X8和X10独立地是C-R4,其中R4是氢或氟,且Q是被取代的或未被取代的杂芳基,条件是当Q是未被取代的杂芳基时,其不是吡啶基,以及当Q是被取代的杂芳基时,其不是6-甲基-3-吡啶基。在本文详述的式(B)的任何实施方案中,在一个实施方案中,Q是被烷基、全卤代烷基、卤素、羟基或烷氧基所取代的苯基。在另一个实施方案中,Q是被两个或三个选自甲基、全卤代烷基、卤素、羟基和烷氧基的基团所取代的苯基。对Q而言具体的苯基取代基是甲基、三氟甲基、氟、氯、羟基、甲氧基、乙氧基和异丙氧基。当Q是被两个或三个基团所取代的苯基时,所述基团可以是相同或不同的。例如,Q可以是二氟苯基、二氯苯基、二甲氧基苯基、二(三氟甲基)苯基、三氟苯基、(氟)(氯)苯基、(氟)(三氟甲基)苯基、(氯)(三氟甲基)苯基或(氟)(甲氧基)苯基。当式(B)化合物中的Q是被取代的苯基时,所述取代基可位于苯环任何可用的位置。例如,单-取代的苯基可以是在苯基的邻、间或对位被取代。任何可行的苯基取代型式都适用于二-或三-取代的苯基(例如,在邻位和对位、在两个邻位、在两个间位、在间位和对位、在邻、间和对位、在两个邻位和一个对位、在两个邻位和一个间位或在两个间位和一个对位或邻位)。在本文详述的式(B)的任何实施方案中,在一个实施方案中,Q是未被取代的吡咯基或被烷基(例如,甲基、乙基或丙基,其包括异丙基)取代的吡咯基。在一个实施方案中,未被取代的吡咯基或被单个基团取代的吡咯基经由该未被取代的吡咯基或被单取代的吡咯基的1、2或3-位连接于带有R8e和R8f的碳。在另一个实施方案中,化合物是式(B)化合物且Q是:(i)4-三氟甲基-3-吡啶基;(ii)未被取代的苯基,条件是X7-X10中至少一个不是CH;或(iii)Q是哌啶基且R8e和R8f与它们所连接的碳一起形成羰基。当可适用时,涉及式(A)和/或式(E)的所有实施方案可同样应用于式B,就像每一个实施方案都具体地和逐一地列出一样。同样,当可适用时,涉及式(A)和/或式(E)的所有实施方案可同样应用于本文详述的任何其他的式,就像每一个实施方案都具体地和逐一地列出一样。在另一个实施方案中,化合物是式(E)或(B)化合物,其中:m是1且q是0(当实施方案涉及式(E)时);R1是CH3或乙基;R8c和R8d各自都是H;R8e和R8f独立地是H、OH或CH3;X7是CH;X8是CH或C-卤素;X10是CH或C-卤素;X9是CR4,其中R4是卤素、CH3、CF3或乙基;且Q是被取代的或未被取代的苯基或被取代的或未被取代的吡啶基,条件是:(i)当R1是CH3,R8e和R8f都是H,X9是CR4,其中R4是F或CF3,且Q是苯基时,该化合物不是244x和245x;或(ii)当R1是CH3,R8e和R8f都是H;且Q是2-吡啶基、2-甲基-3-吡啶基或6-甲基-3-吡啶基时,该化合物不是181x、250x、254x、125x、131x、132x、133x、134x、140x、141x、148x、168x、169x、170x、171x、172x、173x、174x、175x和255x。在一个此类实施方案中,Q是被一个、两个或三个选自卤素、C1-4烷基、-O-C1-4烷基、全卤代烷基和全卤代烷氧基的基团所取代的苯基。在一个实施方案中,Q是被一个、两个或三个选自F、Cl、I、Br、甲基、乙基、丙基、CF3、OMe、OEt和O-CH(CH3)2的基团所取代的苯基。在另一个此类实施方案中,Q是2-吡啶基、3-吡啶基或4-吡啶基,其各自可被一个、两个或三个选自C1-4烷基、卤素、全卤代烷基或全卤代烷氧基的基团所取代。在一个实施方案中,Q是被一个、两个或三个选自F、Cl、I、Br、甲基、乙基、丙基、异-丙基和CF3的基团所取代的吡啶基。本发明还包括式(C)化合物:其中:R1是H、羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R2a和R2b一起形成羰基部分;R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基或硝基或R3a和R3b一起形成羰基部分;X7、X8、X9和X10各自独立地是N或CR4;R4各自独立地是H、羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、羰基烷氧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基;R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H、羟基、C1-C8烷基或与其所连接的碳以及一个孪位的R8一起形成环烷基部分或羰基部分;R10a和R10b各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、羟基、烷氧基或R10a和R10b一起形成羰基;且Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基,条件是该化合物不是化合物202x、205x、206x、207x、208x、209x、210x、211x、212x、213x、214x和221x中的任何一个或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式C化合物,其包括表1中所列的那些化合物,诸如化合物202x、205x、206x、207x、208x、209x、210x、211x、212x、213x、214x和221x,或其盐。在一个实施方案中,化合物是式(C)化合物,条件是(i)当Q是被取代的哌嗪基时,X7、X8、X9和X10中至少一个不是CH;且(ii)该化合物不是化合物202x、214x和221x中的任何一个。在另一个实施方案中,化合物是式C化合物,其中Q是被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基,条件是:(i)当Q是4-(2-甲氧基苯基)哌嗪-1-基时,R1不是H、苄基、甲基、乙酰基或苯甲酰基;或(ii)当Q是4-苄基哌嗪-1-基时,R1不是甲基;或(iii)当Q是4-甲基哌嗪-1-基时,R1不是甲基、苄基或苯乙基;或(iv)所述化合物不是2,3,4,5-四氢-2-甲基-5-[3-(4-吗啉基)丙基]-1H-吡啶并[4,3-b]吲哚,或其盐或溶剂化物。在一个实施方案中,化合物是式C化合物,其中Q是碳环,诸如5、6或7元的碳环。在另一个实施方案中,化合物是式C化合物,其中Q是杂环,诸如5、6或7元的杂环。在另一个实施方案中,化合物是式C化合物,其中Q是被取代的或未被取代的芳基,诸如5、6或7元的芳基。在另一个实施方案中,化合物是式C化合物,其中Q是被取代的或未被取代的杂芳基,诸如5、6或7元的杂芳基。本发明还包括式D化合物:其中:R1是H、羟基、硝基、氰基、卤素、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、全卤代烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基、C1-C8全卤代烷氧基、烷氧基、芳氧基、羧基、硫羟、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基或羰基亚烷基烷氧基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、羟基、烷氧基、氰基、硝基或R2a和R2b一起形成羰基部分;R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基或硝基或R3a和R3b一起形成羰基部分;X7、X8、X9和X10各自独立地是N或CR4;R4各自独立地是H、羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、羰基烷氧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基;R8e和R8f各自独立地是H、羟基、C1-C8烷基或与其所连接的碳及一个孪位的R8一起形成环烷基部分或羰基部分;R10a和R10b各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、羟基、烷氧基或R10a和R10b一起形成羰基;且Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;条件是该化合物不是化合物1x、2x、3x、4x、5x、6x、7x、8x、9x、10x、11x、12x、13x、14x、15x、16x、17x、18x、19x、20x、21x、22x、23x、24x、25x、26x、27x、28x、29x、30x、31x、32x、33x、34x、35x、36x、37x、38x、39x、40x、41x、42x、43x、44x、45x、46x、47x、48x、49x、50x、51x、52x、53x、54x、55x、56x、57x、58x、59x、60x、61x、62x、65x、66x、67x、68x、69x、70x、71x、73x、74x、77x、78x、79x、80x、81x、82x、83x、84x、85x、88x、89x、90x、91x、92x、93x、94x、95x、96x、97x、98x、100x、101x、102x、103x、104x、105x、106x、107x、109x、110x、111x、113x、114x、115x、116x、117x、118x、119x、120x、121x、122x、123x、124x、216x、217x、218x、219x、220x、230x、231x、232x、236x、238x、239x、240x、259x、260x、261x、262x、263x、264x、265x、266x、267x、268x、269x、270x、271x、272x、273x、274x、275x、276x、277x、278x、279x、280x、281x、282x、283x、284x、285x、286x、287x、288x、289x、290x、291x、292x、293x、294x、295x、296x、297x、298x、299x、300x、301x、302x、303x、304x、305x、306x、307x、308x、309x、310x、311x、312x、313x、314x、315x、316x、317x和318x中的任何一个或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式D化合物,其包括表1和表1A中所列那些化合物,诸如化合物1x-62x、65x-71x、73x、74x、77x-85x、88x-98x、100x-107x、109x-111x、113x-124x、216x-220x、230x-232x、236x、238x-240x、259x-295x和296x-318x或其盐。在一个实施方案中,化合物是式D化合物,其中Q是碳环或杂环,诸如5、6或7元的碳环或杂环。在又一个实施方案中,化合物是式D化合物,其中Q是被取代的或未被取代的芳基或被取代的或未被取代的杂芳基,条件是:(i)当Q是被取代的或未被取代的杂芳基时,该化合物不是1-[[2-环戊基-2,3,4,5-四氢-5-[(2-甲基-4-噻唑基)甲基]-1H-吡啶并[4,3-b]吲哚-8-基]羰基]-4-甲基-哌啶;4-甲基-1-[[2,3,4,5-四氢-5-[(2-甲基-4-噻唑基)甲基]-1H-吡啶并[4,3-b]吲哚-8-基]羰基]-哌啶;5-[[2-环戊基-1,2,3,4-四氢-8-[(4-甲基-1-哌啶基)羰基]-5H-吡啶并[4,3-b]吲哚-5-基]甲基]-2-呋喃甲酸;1-[[2-环戊基-2,3,4,5-四氢-5-[(5-甲基-4-异唑基)甲基]-1H-吡啶并[4,3-b]吲哚-8-基]羰基]-4-甲基-哌啶;4-甲基-1-[[2,3,4,5-四氢-5-[(5-甲基-4-异唑基)甲基]-1H-吡啶并[4,3-b]吲哚-8-基]羰基]-哌啶;8-氯-2-甲基-5-(1-(6-甲基吡啶-2-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;8-溴-2-甲基-5-(1-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;8-氯-2-甲基-5-(1-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;且(ii)当Q是被取代的或未被取代的芳基时,(a)其不是5-(2,4-二甲基苄基)-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;5-(4-氯苄基)-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;7-氯-5-(4-氯苄基)-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;8-氯-5-(4-氯苄基)-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;8-氟-5-(4-氟苄基)-2-(2-(吡啶-2-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;(5-(4-羟基苄基)-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-基)(4-羟基苯基)甲酮;和8-氯-5-(4-甲氧基苄基)-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚;或(b)其不是未被取代的苯基;且(iii)当X9是CR4时,R4不是4-甲基哌啶-1-羰基、4-甲氧基哌啶-1-羰基、4-乙氧基哌啶-1-羰基;或其盐或溶剂化物。在一个实施方案中,化合物是式(E)或(D)化合物,条件是当化合物是式(E)化合物时,m和q都是0,且(i)-(vii)中至少一项适用:(i)当X8和X10都是CH,X7是CR4,其中R4是F、CF3或羰基烷氧基,X9是CR4,其中R4是H、甲基、氟或羰基烷氧基,且R1是H或被取代的或未被取代的C1-C5烷基,那么Q不是未被取代的苯基、4-氟苯基、4-乙氧基羰基苯基和未被取代的吡啶基;(ii)当X8和X10都是CH,X7是CR4,其中R4是F、CF3或羰基烷氧基,X9是CR4,其中R4是H、甲基、氟或羰基烷氧基,且R1是羰基烷氧基,那么Q不是苯基、4-氟苯基和未被取代的吡啶基;(iii)当X7-X10各自是CH且R1是羰基烷氧基,那么Q不是苯基和未被取代的吡啶基;(iv)当X8和X10都是CH,X7和X9中的一个是CR4,其中R4是氟、CF3、羧基或羰基烷氧基,且另一个是CH,R1是H、甲基、被取代的C1-C2烷基或羰基烷氧基,那么Q不是苯基、4-氟苯基和未被取代的吡啶基;(v)当X7-X10各自是CH且R1是甲基或丁基,那么Q不是苯基、4-氟苯基和未被取代的吡啶基;(vi)当X7、X8和X10各自是CH;X9是CR4,其中R4是氟、甲基或CF3,且R1是甲基,那么Q不是苯基、被取代的苯基、未被取代的吡啶基、1,2,5,6-四氢吡啶-3-基和2-噻吩基;和(vii)当X8-X10各自是CH、X7是CR4,其中R4是氟或CF3,且R1是甲基,那么Q不是苯基和未被取代的吡啶基。在一个实施方案中,本发明化合物是式(I)化合物,其中:R1是被取代的或未被取代的C1-C8烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基或被取代的或未被取代的芳基;R2a和R2b各自独立地是H、甲基、氟或R2a和R2b一起形成羰基;R3a和R3b各自独立地是H或氟;且R10a和R10b各自独立地是H、卤素、羟基或甲基或R10a和R10b一起形成羰基。式(I)的该实施方案在本文中被称为式“(Ia)”。在一个实施方案中,化合物是式(Ia)化合物,条件是该化合物不是表1中的化合物或其盐。在另一个实施方案中,化合物是式Ia化合物,条件是该化合物不是化合物1x、2x、3x、4x、5x、6x、7x、8x、9x、10x、11x、12x、13x、14x、15x、16x、17x、18x、19x、20x、21x、22x、23x、24x、25x、26x、27x、28x、29x、30x、31x、32x、33x、34x、35x、36x、37x、38x、39x、40x、41x、42x、43x、44x、45x、46x、47x、48x、49x、50x、51x、52x、53x、54x、55x、56x、57x、58x、59x、60x、61x、62x、65x、66x、67x、68x、69x、71x、72x、73x、74x、77x、78x、80x、81x、82x、83x、84x、85x、89x、90x、91x、92x、93x、94x、95x、96x、97x、98x、100x、101x、102x、103x、104x、105x、106x、107x、109x、110x、111x、113x、114x、115x、116x、117x、118x、119x、120x、121x、122x、123x、124x、125x、126x、127x、128x、129x、130x、131x、132x、133x、134x、135x、136x、137x、138x、139x、140x、141x、142x、143x、144x、145x、146x、148x、149x、150x、151x、152x、155x、157x、158x、159x、160x、161x、162x、163x、164x、165x、166x、167x、168x、169x、170x、171x、172x、173x、174x、175x、176x、177x、178x、179x、181x、182x、183x、184x、185x、186x、187x、188x、189x、190x、191x、192x、193x、194x、195x、196x、197x、198x、199x、200x、201x、202x、203x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、214x、219x、220x、221x、222x、223x、224x、225x、226x、227x、228x、229x、230x、231x、232x、233x、234x、235x、236x、237x、238x、239x、243x、244x、245x、246x、247x、248x、249x、250x、251x、252x、253x、254x、255x、256x、257x、258x、259x、260x、261x、262x、263x、264x、265x、266x、267x、268x、269x、270x、271x、272x、273x、274x、275x、276x、277x、278x、279x、280x、281x、282x、283x、284x、285x、286x、287x、288x、289x、290x、291x、292x、293x、294x和295x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式Ia化合物,其包括表1中列出的那些化合物或其盐。当可适用时,涉及式(I)的所有实施方案可同样应用于式A-F,就像每一个实施方案都具体地和逐一地列出一样。同样,当可适用时,涉及式(I)的所有实施方案可同样应用于本文详述的所有的式和实施方案,就像每一个实施方案都具体地和逐一地列出一样。在一个特定的实施方案中,化合物是式(I)或Ia化合物,其中X7、X8、X9和X10是CR4。在另一个实施方案中,化合物是式(I)或Ia化合物,其中X7、X8、X9和X10中至少一个是N。另一个实施方案提供了式(I)或Ia化合物,其中X7、X8、X9和X10中至少两个是N。另一个实施方案提供了式(I)或Ia化合物,其中X7、X8、X9和X10中两个是N且X7、X8、X9和X10中两个是CR4。本发明还包括式(I)或Ia化合物,其中X7、X8、X9和X10中的一个是N且X7、X8、X9和X10中的三个是CR4。在另一个实施方案中,本发明化合物是式(I)或Ia化合物,其中X7、X8、X9和X10一起提供选自以下结构的芳族基团:其中R4各自如对式(I)或Ia所定义;或在一个特定的实施方案中,其中R4各自独立地是羟基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、烷基磺酰基氨基或酰基;或在又一个实施方案中,其中R4独立地是卤素、未被取代的C1-C4烷基或C1-C4全卤代烷基。在一个实施方案中,R4是羟基、羧基、甲基、-CF3、氯、-OCF3、异-丙基、乙基、叔-丁基、-CH2OH、溴、碘、氟、3-吡啶基或-CH2CH2CH2NH2。在一个实施方案中,R4不是氢。在又一个实施方案中,本发明化合物是式(I)或Ia化合物,其中X7、X8、X9和X10一起提供自以下结构的芳族基团:其中R4是如在式(I)中所定义;或在一个特定的实施方案中,其中R4是羟基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C1-C8全卤代烷氧基、C1-C8烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、烷基磺酰基氨基或酰基;或在又一个实施方案中,其中R4各自独立地是卤素、未被取代的C1-C4烷基或C1-C4全卤代烷基。在又一个实施方案中,R4是H、羟基、羧基、甲基、-CF3、氯、-OCF3、异-丙基、乙基、叔-丁基、-CH2OH、溴、碘、氟、3-吡啶基或-CH2CH2CH2NH2。在另一个实施方案中,本发明化合物是式(I)或Ia化合物或任何本文的实施方案,其中X7、X8、X9和X10一起形成其中R4如在本文的任何实施方案中所定义,包括其中R4是H、羟基、羧基、甲基、-CF3、氯、-OCF3、异-丙基、乙基、叔-丁基、-CH2OH、溴、碘、氟、3-吡啶基或-CH2CH2CH2NH2。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10如在式(I)中所定义或如在本文任何实施方案中详述,其中R1是H、被取代的或未被取代的C1-C8烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的芳烷基。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10如在式(I)中所定义或如在本文任何实施方案中详述,其中R1是被取代的或未被取代的C1-C8烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基或被取代的或未被取代的芳基。在一个特定的实施方案中,本发明化合物是式(I)化合物,其中X7-X10如在式(I)中所定义或如在本文任何实施方案中详述,其中R1是甲基、乙基、环丙基、丙醇基(propylate)、三氟甲基、异丙基、叔-丁基、仲-丁基、2-甲基丁基、丙醛、1-甲基-2-羟基乙基、2-羟基乙醛、2-羟基乙基、2-羟基丙基、2-羟基-2-甲基丙基、环丁基、环戊基、环己基、被取代的苯基、哌啶-4-基、羟基环戊-3-基、羟基环戊-2-基、羟基环丙-2-基、1-羟基-1-甲基环丙-2-基或1-羟基-1,2,2-三甲基-环丙-3-基。在本文任何通式或子结构的另一个实施方案中,R1是H、羧基、丙基、乙氧基羰基、2,2,2-三氯乙氧基羰基、羟基甲基、3-氨基-丙基、甲酰基、正-丁基、苯基或中任何一种。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a和R2b独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基、硝基或R2a和R2b一起形成羰基部分,且R3a和R3b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基或硝基。在另一个实施方案中,化合物是式(I)化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a和R2b各自独立地是H、未被取代的C1-C8烷基、卤素或R2a和R2b一起形成羰基部分,且R3a和R3b各自独立地是H、未被取代的C1-C8烷基、卤素或R3a和R3b一起形成羰基部分。在又一个实施方案中,化合物是式(I)化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a和R2b各自独立地是H、未被取代的C1-C8烷基、卤素或R2a和R2b一起形成羰基部分;且R3a和R3b各自独立地是H、未被取代的C1-C8烷基、卤素或R3a和R3b一起形成羰基部分。本发明还包括式(I)的本发明化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a和R2b各自独立地是H、甲基、卤素或R2a和R2b一起形成羰基部分,且R3a和R3b各自独立地是H、甲基、卤素或R3a和R3b一起形成羰基部分。本发明进一步包括式(I)的本发明化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a、R2b、R3a和R3b各自是H。在一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a、R2b、R3a和R3b中至少一个是被取代的或未被取代的C1-C8烷基、卤素、氰基、硝基或与一个孪位的R2或R3一起形成羰基部分。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a、R2b、R3a和R3b中至少两个是被取代的或未被取代的C1-C8烷基、卤素、氰基、硝基或与一个孪位的R2或R3一起形成羰基部分。在又一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a、R2b、R3a和R3b中至少一个是氟或甲基或与一个孪位的R2或R3一起形成羰基部分。在又一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10和R1如在式(I)中所定义或如在本文任何实施方案中详述,其中R2a和R2b、或者R3a和R3b各自是甲基或氟(例如,R2a和R2b都是甲基或一个是氟且一个是甲基)或一起形成羰基部分。在一个实施方案中,R2a和R2b一起形成羰基部分。在另一个实施方案中,R2a和R2b中至少一个是羟基或烷氧基。在一个特定的实施方案中,R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基、硝基或R2a和R2b一起形成羰基。在另一个实施方案中,当X1是N时,R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基、卤素、氰基、硝基或R2a和R2b一起形成羰基。本发明还包括式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a和R10b各自独立地是H、卤素、未被取代的C1-C8烷基、羟基或R10a和R10b一起形成羰基。还包括式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a和R10b各自独立地是H、卤素、未被取代的C1-C4烷基、羟基或R10a和R10b一起形成羰基。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a和R10b各自独立地是H、溴、甲基、羟基或R10a和R10b一起形成羰基。在又一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a和R10b中至少一个是未被取代的C1-C8烷基、羟基、卤素或R10a和R10b一起形成羰基。在又一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a和R10b中至少一个是甲基、溴、羟基或R10a和R10b一起形成羰基。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a和R10b都是甲基。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a和R10b一起形成羰基。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a是H且R10b是甲基。在另一个实施方案中,本发明化合物是式(I)化合物,其中X7-X10、R1、R2a、R2b、R3a和R3b如在式(I)中所定义或如在本文任何实施方案中详述,其中R10a是H且R10b是溴。当带有R10a和R10b的式(I)的碳具有旋光性,其可以是S或R构型,且包含基本上纯的R或S化合物或其以任何的量的混合物的组合物也包括在本发明内。在一个特定的实施方案中,本发明化合物是式(I)化合物,其中R2a、R2b、X1、R10a、R10b、R3a和R3b一起形成选自以下结构的环:其中以上结构中的R1如对式(I)或本文详述的任何特定的实施方案所定义。在另一个实施方案中,本发明包括具有R2a、R2b、X1、R10a,R10b、R3a和R3b的化合物,其中取代基R2a、R2b、X1、R10a,R10b、R3a和R3b一起形成下式的基团:在一个特定的实施方案中,本文详述的任何结构或子结构的R1是甲基。本发明还包括式(IIa)、(IIb)、(IIc)、(IId)、(IIe)、(IIf)、(IIg)和(IIh)的化合物:其中在(IIa)、(IIb)、(IIc)、(IId)、(IIe)、(IIf)、(IIg)和(IIh)中的每一个中,R1、R2a、R2b、R3a、R3b,R10a,R10b、R8a-R8f、m、q和Q如对式(I)或其任何可适用的实施方案所定义。当可适用时,在(IIa)、(IIb)、(IIc)、(IId)、(IIe)、(IIf)、(IIg)和(IIh)中的每一个中,R1、R2a、R2b、R3a、R3b、R10a、R10b、R8a-R8f、m、q和Q还可如对本文详述的任何式或其任何可适用的实施方案所描述,其包括但不限于式(A)-(G)。在一个实施方案中,化合物是式(IIa)化合物,条件是当X1是N时,该化合物不是化合物13x、30x、45x、51x、53x、54x、55x、57x、58x、61x、62x、65x、68x、70x、72x、77x、78x、79x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、135x、138x、140x、142x、143x、144x、145x、146x、156x、157x、160x、161x、163x、166x、177x、179x、180x、181x、182x、183x、190x、192x、195x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、216x、217x、221x、222x、223x、225x、226x、227x、228x、236x、237x、248x、259x、260x、261x、266x、267x和268x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(IIa)化合物,其包括表1中所列的那些化合物,诸如化合物13x、30x、45x、51x、53x-55x、57x、58x、61x、62x、65x、68x、70x、72x、77x-79x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、135x、138x、140x、142x-146x、156x、157x、160x、161x、163x、166x、177x、179x-183x、190x、192x、195x、202x、204x-213x、216x、217x、221x-223x、225x-228x、236x、237x、248x、259x、260x、261x、266x、267x和268x或其盐。在一个实施方案中,化合物是式(IIa)化合物,其中q是0,R1是甲基或乙基且Q是被取代的苯基。在一个实施方案中,化合物是式(IId)化合物,条件是该化合物不是化合物233x、234x和235x中的任何一种。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(IId)化合物,其包括表1中所列的那些化合物,诸如化合物233x、234x和235x。在另一个实施方案中,化合物是式(IId)化合物,条件是当Q是被取代的哌嗪基时,R8e和R8f不与它们所连接的碳一起形成羰基。在又一个实施方案中,化合物是式(IId)化合物,条件是当Q是被取代的哌嗪基且R8e和R8f与它们所连接的碳一起形成羰基时,R1不是异丙基。在又一个实施方案中,化合物是式(IId)化合物,其中R1是甲基。在一个特定的实施方案中,化合物是式(IId)化合物,其中R1是甲基且q是0。在又一个实施方案中,化合物是式(IId)化合物,其中R1是甲基,且R2a、R2b、R3a、R3b、R10a、R10b及R8a-R8f中的每一个,当存在时,是氢。本发明进一步包括式(IIIa)、(IIIb)、(IIIc)、(IIId)、(IIIe)、(IIIf)、(IIIg)、(IIIh)、(IIIi)、(IIIj)、(IIIk)、(IIIl)、(IIIm)的化合物:其中在(IIIa)、(IIIb)、(IIIc)、(IIId)、(IIIe)、(IIIf)、(IIIg)、(IIIh)、(IIIi)、(IIIj)、(IIIk)、(IIIl)和(IIIm)中的每一个中,R1、R4、R2a、R2b、R3a、R3b、R10a、R10b、R8a-R8f、m、q和Q如对式(I)或其任何可适用的实施方案所描述。当可适用时,在(IIIa)、(IIIb)、(IIIc)、(IIId)、(IIIe)、(IIIf)、(IIIg)、(IIIh)、(IIIi)、(IIIj)、(IIIk)、(IIIl)和(IIIm)中的每一个中,R1、R4、R2a、R2b、R3a、R3b、R10a、R10b、R8a-R8f、m、q和Q还可如对本文详述的任何式或其任何可适用的实施方案所描述,其包括但不限于式(A)-(G)。在一个实施方案中,化合物是式(IIIa)化合物,条件是(i)当X1是N时,该化合物不是化合物1x、2x、3x、4x、5x、6x、7x、8x、9x、10x、11x、12x、13x、15x、16x、17x、18x、19x、20x、21x、22x、23x、24x、25x、26x、27x、28x、29x、30x、31x、32x、33x、34x、35x、36x、37x、38x、39x、40x、41x、42x、43x、44x、45x、47x、49x、50x、51x、52x、53x、54x、55x、56x、57x、58x、59x、60x、61x、62x、65x、68x、69x、70x、72x、77x、78x、79x、81x、84x、85x、88x、89x、90x、92x、93x、94x、95x、96x、97x、98x、100x、101x、102x、103x、104x、105x、106x、107x、109x、110x、111x、115x、116x、117x、118x、119x、120x、121x、122x、123x、124x、125x、126x、127x、128x、129x、130x、134x、135x、136x、137x、138x、139x、140x、142x、143x、144x、145x、146x、148x、149x、150x、151x、152x、155x、156x、157x、158x、159x、160x、161x、162x、163x、164x、165x、166x、167x、171x、174x、175x、176x、177x、178x、179x、180x、181x、182x、183x、184x、189x、190x、191x、192x、193x、194x、195x、196x、198x、199x、200x、201x、202x、203x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、214x、216x、217x、219x、220x、221x、222x、223x、224x、225x、226x、227x、228x、229x、231x、232x、236x、237x、238x、239x、243x、244x、245x、248x、249x、250x、251x、252x、253x、254x、255x、256x、258x、259x、260x、261x、262x、263x、264x、265x、266x、267x、268x、269x、270x、271x、272x、273x、274x、275x、276x、279x、280x、281x、282x、285x、286x、287x、288x、290x、291x、292x、293x、294x和295x中的任何一种或其盐且(ii)该化合物不是化合物229H、230、241、255、256、262和274中的任何一种或其盐。在另一个实施方案中,本发明化合物与本文详述的使用该化合物及施用所述化合物的方法,包括任何式(IIIa)化合物,其包括表1中所列的那些化合物,诸如化合物1x-13x、15x-45x、47x、49x-62x、65x、68x-70x、72x、77x-79x、81x、84x、85x、88x-90x、92x-98x、100x-107x、109x-111x、115x-130x、134x-140x、142x-146x、148x-152x、155x-167x、171x、174x-184x、189x-196x、198x-214x、216x、217x、219x-229x、231x、232x、236x-239x、243x-245x、248x-256x、258x-276x、279x-282x、285x-288x和290x-295x或其盐及化合物229H、230、241、255、256、262和274或其盐。在另一个实施方案中,化合物是式(IIIa)化合物,其中R4不是H,R1是甲基,m是0且q是1,且其中Q是被取代的或未被取代的芳基、被取代的或未被取代的C3-8环烷基或被取代的或未被取代的C3-8环烯基。在一个特定的实施方案中,化合物是式(IIIa)化合物,其中R4不是氢;q是0且(i)-(iii)中至少一项适用:(i)R8c、R8d、R8e和R8f中至少一个是羟基、C1-C8烷基、C1-C8全卤代烷基、羧基、羰基烷氧基或与其连接的碳及一个孪位的R8一起形成环烷基部分或羰基部分;(ii)X7-X10中至少一个是N;(iii)m是0,R8c、R8d、R8e和R8f各自是氢且Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基,条件是:(a)当Q是未被取代的杂芳基时,其不是吡啶基;(a)当Q是被取代的杂芳基时,其不是6-甲基-3-吡啶基;且(c)当Q是未被取代的杂环基,其是具有至少两个环杂原子的未被取代的杂环基。在一个实施方案中,化合物是式(IIIb)化合物,条件是当X1是N时,该化合物不是化合物13x、30x、45x、51x、53x、54x、55x、57x、58x、61x、62x、65x、66x、67x、68x、70x、72x、77x、78x、79x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、135x、138x、140x、142x、143x、144x、145x、146x、156x、157x、160x、161x、163x、166x、177x、179x、180x、181x、182x、183x、190x、192x、195x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、216x、217x、221x、222x、223x、225x、226x、227x、228x、236x、237x、248x、259x、260x、261x、266x、267x和268x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(IIIb)化合物,其包括表1中所列的那些化合物,诸如化合物13x、30x、45x、51x、53x-55x、57x、58x、61x、62x、65x-68x、70x、72x、77x-79x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、135x、138x、140x、142x-146x、156x、157x、160x、161x、163x、166x、177x、179x-183x、190x、192x、195x、202x、204x-213x、216x、217x、221x-223x、225x-228x、236x、237x、248x、259x、260x、261x、266x、267x和268x或其盐。在一个实施方案中,化合物是式(IIIb)化合物,其中R4不是氢,条件是该化合物不是66x或67x。在另一个实施方案中,化合物是式(IIIb)化合物,其中m是0,q是1且R4是羟基、硝基、氰基、卤素、C1-C8全卤代烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、被取代的或未被取代的芳基、被取代的或未被取代的杂芳基、C2-C8全卤代烷氧基、C1-C8烷氧基、芳氧基、羧基、羰基烷氧基、硫羟、被取代的或未被取代的杂环基、被取代的或未被取代的芳烷基、烷硫基、被取代的或未被取代的氨基、酰基氨基、氨基酰基、氨基羰基氨基、氨基羰基氧基、氨基磺酰基、磺酰基氨基、磺酰基、羰基亚烷基烷氧基、烷基磺酰基氨基或酰基。在一个实施方案中,化合物是式(IIIc)化合物,条件是当X1是N时,该化合物不是化合物13x、14x、30x、45x、51x、53x、54x、55x、57x、58x、61x、62x、65x、68x、70x、72x、77x、78x、79x、80x、82x、83x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、132x、135x、138x、140x、141x、142x、143x、144x、145x、146x、156x、157x、160x、161x、163x、166x、170x、177x、179x、180x、181x、182x、183x、187x、190x、192x、195x、197x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、216x、217x、221x、222x、223x、225x、226x、227x、228x、230x、236x、237x、248x、259x、260x、261x、266x、267x和268x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(IIIc)化合物,其包括表1中所列的那些化合物,诸如化合物13x、14x、30x、45x、51x、53x-55x、57x、58x、61x、62x、65x、68x、70x、72x、77x-80x、82x、83x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、132x、135x、138x、140x-146x、156x、157x、160x、161x、163x、166x、170x、177x、179x-183x、187x、190x、192x、195x、197x、202x、204x-213x、216x、217x、221x-223x、225x-228x、230x、236x、237x、248x、259x-261x和266x-268x或其盐。在一个实施方案中,化合物是式(IIIc)化合物,其中X1是N且R4不是氢,条件是该化合物不是化合物14x、80x、82x、83x、132x、141x、170x、187x、197x或230x。在一个实施方案中,化合物是式(IIIc)化合物,其中X1是N、R4不是氢,m是0且q是1,R8e和R8f都是H且Q不是6-甲基-3-吡啶基或4-吡啶基。在一个实施方案中,化合物是式(IIId)化合物,条件是当X1是N时,该化合物不是化合物13x、30x、45x、46x、51x、53x、54x、55x、57x、58x、61x、62x、65x、68x、70x、71x、72x、77x、78x、79x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、135x、138x、140x、142x、143x、144x、145x、146x、156x、157x、160x、161x、163x、166x、177x、179x、180x、181x、182x、183x、185x、190x、192x、195x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、216x、217x、218x、221x、222x、223x、225x、226x、227x、228x、236x、237x、246x、247x、248x、257x、259x、260x、261x、266x、267x、268x、277x、278x、283x、284x和289x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(IIId)化合物,其包括表1中所列的那些化合物,诸如化合物13x、30x、45x、46x、51x、53x-55x、57x、58x、61x、62x、65x、68x、70x-72x、77x-79x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、135x、138x、140x、142x-146x、156x、157x、160x、161x、163x、166x、177x、179x-183x、185x、190x、192x、195x、202x、204x-213x、216x-218x、221x-223x、225x-228x、236x、237x、246x-248x、257x、259x-261x、266x-268x、277x、278x、283x、284x和289x或其盐。在又一个实施方案中,化合物是式(IIId)化合物,其中R4不是氢,条件是该化合物不是化合物46x、71x、185x、218x、246x、247x、257x、277x、278x、283x、284x和289x中的任何一种。在又一个实施方案中,化合物是式(IIId)化合物,其中R4不是氢,m是0且q是1且Q不是6-甲基-3-吡啶基和4-吡啶基。在又一个实施方案中,化合物是式(III)化合物,其中R4是卤素或羟基,m是0且q是1。在一个实施方案中,化合物是式(IIIe)化合物,条件是(i)当X1是N时,该化合物不是化合物1x、2x、3x、4x、5x、6x、7x、8x、9x、10x、11x、12x、13x、14x、15x、16x、17x、18x、19x、20x、21x、22x、23x、24x、25x、26x、27x、28x、29x、30x、31x、32x、33x、34x、35x、36x、37x、38x、39x、40x、41x、42x、43x、44x、45x、47x、49x、50x、51x、52x、53x、54x、55x、56x、57x、58x、59x、60x、61x、62x、65x、68x、69x、70x、72x、77x、78x、79x、80x、81x、82x、83x、84x、85x、88x、89x、90x、92x、93x、94x、95x、96x、97x、98x、100x、101x、102x、103x、104x、105x、106x、107x、109x、110x、111x、113x、115x、116x、117x、118x、119x、120x、121x、122x、123x、124x、125x、126x、127x、128x、129x、130x、131x、132x、134x、135x、136x、137x、138x、139x、140x、141x、142x、143x、144x、145x、146x、148x、149x、150x、151x、152x、155x、156x、157x、158x、159x、160x、161x、162x、163x、164x、165x、166x、167x、168x、169x、170x、171x、173x、174x、175x、176x、177x、178x、179x、180x、181x、182x、183x、184x、186x、187x、189x、190x、191x、192x、193x、194x、195x、196x、197x、198x、199x、200x、201x、202x、203x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、214x、216x、217x、219x、220x、221x、222x、223x、224x、225x、226x、227x、228x、229x、230x、231x、232x、236x、237x、238x、239x、243x、244x、245x、248x、249x、250x、251x、252x、253x、254x、255x、256x、258x、259x、260x、261x、262x、263x、264x、265x、266x、267x、268x、269x、270x、271x、272x、273x、274x、275x、276x、279x、280x、281x、282x、285x、286x、287x、288x、290x、291x、292x、293x、294x和295x中的任何一种或其盐且(ii)该化合物不是化合物229H、230、241、255、256、262和274中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括任何式(IIIe)化合物,其包括表1中所列的那些化合物,诸如化合物1x-45x、47x、49x-62x、65x、68x-70x、72x、77x-85x、88x-90x、92x-98x、100x-107x、109x-111x、113x、115x-132x、134x-146x、148x-152x、155x-171x、173x-184x、186x、187x、189x-214x、216x、217x、219x-232x、236x-239x、243x-245x、248x-256x、258x-276x、279x-282x、285x-288x和290x-295x或其盐以及化合物229H、230、241、255、256、262和274或其盐。在另一个实施方案中,化合物是式(IIIe)化合物,其中R4各自不是氢,条件是该化合物不是113x、131x、168x、169x、173x或186x。在另一个实施方案中,化合物是式(IIIe)化合物,其中R4各自不是氢,R8e和R8f都是氢且Q不是4-吡啶基或6-甲基-3-吡啶基。在一个实施方案中,化合物是式(IIIf)化合物,其中该化合物不是化合物233x、234x和235x中的任何一种。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括任何式(IIIf)化合物,其包括表1中所列的那些化合物,诸如233x、234x或235x。在另一个实施方案中,本发明化合物是式(IIIf)化合物,条件是当Q是被取代的哌嗪基时,R8e和R8f不与它们所连接的碳一起形成羰基。在又一个实施方案中,本发明化合物是式(IIIf)化合物,其中R4不是氢。在又一个实施方案中,本发明化合物是式(IIIf)化合物,其中R4是甲基或三氟甲基。在另一个此类实施方案中,本发明化合物是式(IIIf)化合物,其中R4是甲基或三氟甲基,R1是甲基且m是0且q是1。在一个实施方案中,化合物是式(IIIk)化合物,其中该化合物不是化合物1x、2x、3x、4x、5x、6x、7x、8x、9x、10x、11x、12x、13x、15x、16x、17x、18x、19x、20x、21x、22x、23x、24x、25x、26x、27x、28x、29x、30x、31x、32x、33x、34x、35x、36x、37x、38x、39x、40x、41x、42x、43x、44x、45x、46x、47x、49x、50x、51x、52x、53x、54x、55x、56x、57x、58x、59x、60x、61x、62x、65x、68x、69x、70x、71x、72x、77x、78x、79x、81x、84x、85x、88x、89x、90x、92x、93x、94x、95x、96x、97x、98x、100x、101x、102x、103x、104x、105x、106x、107x、109x、110x、111x、115x、116x、117x、118x、119x、120x、121x、122x、123x、124x、125x、126x、127x、128x、129x、130x、134x、135x、136x、137x、138x、139x、140x、142x、143x、144x、145x、146x、148x、149x、150x、151x、152x、155x、156x、157x、158x、159x、160x、161x、162x、163x、164x、165x、166x、167x、171x、172x、174x、175x、176x、177x、178x、179x、180x、181x、182x、183x、184x、185x、189x、190x、191x、192x、193x、194x、195x、196x、198x、199x、200x、201x、202x、203x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、214x、216x、217x、218x、219x、220x、221x、222x、223x、224x、225x、226x、227x、228x、229x、231x、232x、236x、237x、238x、239x、243x、244x、245x、246x、247x、248x、249x、250x、251x、252x、253x、254x、255x、256x、257x、258x、259x、260x、261x、262x、263x、264x、265x、266x、267x、268x、269x、270x、271x、272x、273x、274x、275x、276x、277x、278x、279x、280x、281x、282x、283x、284x、285x、286x、287x、288x、289x、290x、291x、292x、293x、294x和295x中的任何一种。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括任何式(IIIk)化合物,其包括表1中所列的那些化合物,诸如化合物1x-13x、15x-47x、49x-62x、65x、68x-72x、77x-79x、81x、84x、85x、88x-90x、92x-98x、100x-107x、109x-111x、115x-130x、134x-140x、142x-146x、148x-152x、155x-167x、171x、172x、174x-185x、189x-196x、198x、199x-214x、216x-229x、231x、232x、236x-239x和243x-295x。在又一个实施方案中,化合物是式(IIIk)化合物,其中R4各自不是氢,条件是该化合物不是化合物172x。在又一个实施方案中,化合物是式(IIIk)化合物,其中R4各自独立地是烷基、羟基或卤素,条件是当一个R4是烷基时,另一个R4是烷基或羟基。在又一个实施方案中,化合物是式(IIIk)化合物,其中R4各自独立地是烷基、羟基或卤素且Q不是6-甲基-3-吡啶基。在一个实施方案中,化合物是式(IIIl)化合物,其中该化合物不是化合物13x、14x、30x、45x、51x、53x、54x、55x、57x、58x、61x、62x、65x、66x、67x、68x、70x、72x、77x、78x、79x、80x、82x、83x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、132x、135x、138x、140x、141x、142x、143x、144x、145x、146x、156x、157x、160x、161x、163x、166x、170x、177x、179x、180x、181x、182x、183x、187x、190x、192x、195x、197x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、216x、217x、221x、222x、223x、225x、226x、227x、228x、230x、236x、237x、248x、259x、260x、261x、266x、267x和268x中的任何一种。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括任何式(IIIl)化合物,其包括表1中所列的那些化合物,诸如化合物13x、14x、30x、45x、51x、53x-55x、57x、58x、61x、62x、65x-68x、70x、72x、77x-80x、82x、83x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、132x、135x、138x、140x-146x、156x、157x、160x、161x、163x、166x、170x、177x、179x-183x、187x、190x、192x、195x、197x、202x、204x-213x、216x、217x、221x-223x、225x-228x、230x、236x、237x、248x、259x-261x和266x-268x。在又一个实施方案中,化合物是式(IIIl)化合物,其中R4各自不是氢。在一个实施方案中,化合物是式(IIIm)化合物,其中该化合物不是化合物13x、30x、45x、46x、48x、51x、53x、54x、55x、57x、58x、61x、62x、65x、66x、67x、68x、70x、71x、72x、73x、74x、77x、78x、79x、88x、89x、91x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、135x、138x、140x、142x、143x、144x、145x、146x、156x、157x、160x、161x、163x、166x、177x、179x、180x、181x、182x、183x、185x、190x、192x、195x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、216x、217x、218x、221x、222x、223x、225x、226x、227x、228x、236x、237x、246x、247x、248x、257x、259x、260x、261x、266x、267x、268x、277x、278x、283x、284x和289x中的任何一种。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括任何式(IIIm)化合物,其包括表1中所列的那些化合物,诸如化合物13x、30x、45x、46x、48x、51x、53x-55x、57x、58x、61x、62x、65x-68x、70x-74x、77x-79x、88x、89x、91x-94x、96x、97x、100x、101x、104x、105x、125x-130x、135x、138x、140x、142x-146x、156x、157x、160x、161x、163x、166x、177x、179x-183x、185x、190x、192x、195x、202x、204x-213x、216x-218x、221x-223x、225x-228x、236x、237x、246x-248x、257x、259x-261x、266x-268x、277x、278x、283x、284x和289x。在又一个实施方案中,化合物是式(IIIm)化合物,其中R4各自不是氢,条件是该化合物不是化合物48x、73x、74x和91x中的任何一种。在又一个实施方案中,化合物是式(IIIm)化合物,其中R4各自不是氢,m是0且q是1。本发明进一步包括式(IVa)、(IVb)、(IVc)、(IVd)、(IVe)、(IVf)、(IVg)、(IVh)、(IVi)、(IVj)和(IVk)的化合物:其中在(IVa)、(IVb)、(IVc)、(IVd)、(IVe)、(IVf)、(IVg)、(IVh)、(IVi)、(IVj)和(IVk)中的每一个中,R1、X7、X8、X9、X10、R8a-R8f、m、q和Q如对式(I)或其任何可适用的实施方案所描述。当可适用时,在(IVa)、(IVb)、(IVc)、(IVd)、(IVe)、(IVf)、(IVg)、(IVh)、(IVi)、(IVj)和(IVk)中的每一个中,R1、X7、X8、X9、X10、R8a-R8f、m、q和Q还可如对本文详述的任何式或其任何可适用的实施方案所描述,其包括但不限于式(A)-(G)。在一个实施方案中,化合物是式(IVa)化合物,条件是(i)该化合物不是化合物1x-44x、47x-50x、52x、54x-62x、65x-67x、71x-74x、77x、78x、80x-85x、88x-98x、100x-107x、109x-111x、113x-146x、148x-152x、155x、157x-179x、181x-214x、219x-239x和243x-295x中的任何一种或其盐且(ii)该化合物不是化合物229H、230、255、256、262和274中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括任何式(IVa)化合物,其包括表1中所列的那些化合物,诸如化合物1x-44x、47x-50x、52x、54x-62x、65x-67x、71x-74x、77x、78x、80x-85x、88x-98x、100x-107x、109x-111x、113x-146x、148x-152x、155x、157x-179x、181x-214x、219x-239x和243x-295x或其盐以及化合物229H、230、255、256、262和274或其盐。在一个实施方案中,化合物是式(IVb)化合物,条件是R1是NRaRb且:(a)Ra和Rb各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,条件是Ra和Rb基团不都是H,且(i)-(iii)中至少一项适用:(i)当m和q是0时,Ra和Rb中至少一个不是甲基;(ii)当m和q是0时,X7-X10中至少一个不是CH;(iii)当m和q是0时,(a)Q不是被取代的或未被取代的苯基或(b)Q是被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基;或(b)Ra和Rb与氮原子一起形成杂环或被取代的杂环。在式(IVb)的一个实施方案中,m和q中至少一个是1,R1是NRaRb,且(a)Ra和Rb基团各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,条件是Ra和Rb基团不都是H,或(b)Ra和Rb与氮原子一起形成杂环或被取代的杂环。在另一个实施方案中,化合物是(IVj)化合物,条件是该化合物不是化合物45x、46x、51x、216x、217x和218x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(IVj)化合物,其包括表1中所列的那些化合物,诸如45x、46x、51x、216x、217x和218x或其盐。本发明还包括式(Va)-(Vzv)化合物:其中在(Va)-(Vzv)中的每一个中,R1、R4、R8a-R8f、m、q和Q如对式(I)或其任何可适用的实施方案所定义。当可适用时,在(Va)-(Vzv)中的每一个中,R1、R4、R8a-R8f、m、q和Q还可如对本文详述的任何式或其任何可适用的实施方案所描述,其包括但不限于式(A)-(G)。在一个实施方案中,化合物是式(Va)化合物,条件是该化合物不是化合物13x、30x、54x、55x、57x、58x、61x、62x、65x、72x、77x、78x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、135x、138x、140x、142x、143x、144x、145x、146x、157x、160x、161x、163x、166x、177x、179x、181x、182x、183x、190x、192x、195x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、221x、222x、223x、225x、226x、227x、228x、236x、237x、248x、259x、260x、261x、266x、267x和268x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(Va)化合物,其包括表1中所列的那些化合物,诸如化合物13x、30x、54x、55x、57x、58x、61x、62x、65x、72x、77x、78x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、135x、138x、140x、142x-146x、157x、160x、161x、163x、166x、177x、179x、181x-183x、190x、192x、195x、202x、204x-213x、221x-223x、225x-228x、236x、237x、248x、259x、260x、261x、266x、267x和268x或其盐。在一个实施方案中,化合物是式(Vd)化合物,条件是该化合物不是化合物233x、234x和235x中的任何一种。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(Vd)化合物,其包括表1中所列的那些化合物,诸如233x、234x和235x。在另一个实施方案中,化合物是式(Vd)化合物,条件是当Q是被取代的哌嗪基时,R8e和R8f不与它们所连接的碳一起形成羰基。在一个实施方案中,化合物是式(Ve)化合物,条件是该化合物不是化合物13x、30x、54x、55x、57x、58x、61x、62x、65x、66x、67x、72x、77x、78x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、135x、138x、140x、142x、143x、144x、145x、146x、157x、160x、161x、163x、166x、177x、179x、181x、182x、183x、190x、192x、195x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、221x、222x、223x、225x、226x、227x、228x、236x、237x、248x、259x、260x、261x、266x、267x和268x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(Ve)化合物,其包括表1中所列的那些化合物,诸如化合物13x、30x、54x、55x、57x、58x、61x、62x、65x-67x、72x、77x、78x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、135x、138x、140x、142x-146x、157x、160x、161x、163x、166x、177x、179x、181x-183x、190x、192x、195x、202x、204x-213x、221x-223x、225x-228x、236x、237x、248x、259x、260x、261x、266x、267x和268x或其盐。在另一个实施方案中,化合物是式(Ve)化合物,其中R4不是氢,条件是该化合物不是化合物66x和67x中的任何一种。在另一个实施方案中,化合物是式(Ve)化合物,其中R4不是氢(例如,其中R4是氯),m是0且q是1。在一个实施方案中,化合物是式(Vf)化合物,条件是该化合物不是化合物1x、2x、3x、4x、5x、6x、7x、8x、9x、10x、11x、12x、13x、15x、16x、17x、18x、19x、20x、21x、22x、23x、24x、25x、26x、27x、28x、29x、30x、31x、32x、33x、34x、35x、36x、37x、38x、39x、40x、41x、42x、43x、44x、47x、49x、50x、52x、54x、55x、56x、57x、58x、59x、60x、61x、62x、65x、72x、77x、78x、81x、84x、85x、88x、89x、90x、92x、93x、94x、95x、96x、97x、98x、100x、101x、102x、103x、104x、105x、106x、107x、109x、110x、111x、115x、116x、117x、118x、119x、120x、121x、122x、123x、124x、125x、126x、127x、128x、129x、130x、134x、135x、136x、137x、138x、139x、140x、142x、143x、144x、145x、146x、148x、149x、150x、151x、152x、155x、157x、158x、159x、160x、161x、162x、163x、164x、165x、166x、167x、171x、174x、175x、176x、177x、178x、179x、181x、182x、183x、184x、189x、190x、191x、192x、193x、194x、195x、196x、198x、199x、200x、201x、202x、203x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、214x、219x、220x、221x、222x、223x、224x、225x、226x、227x、228x、229x、231x、232x、236x、237x、238x、239x、243x、244x、245x、248x、249x、250x、251x、252x、253x、254x、255x、256x、258x、259x、260x、261x、262x、263x、264x、265x、266x、267x、268x、269x、270x、271x、272x、273x、274x、275x、276x、279x、280x、281x、282x、285x、286x、287x、288x、290x、291x、292x、293x、294x和295x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括任何式(Vf)化合物,其包括表1中所列的那些化合物,诸如化合物1x-13x、15x-44x、47x、49x、50x、52x、54x-62x、65x、72x、77x、78x、81x、84x、85x、88x-90x、92x-98x、100x-107x、109x-111x、115x-130x、134x-140x、142x-146x、148x-152x、155x、157x-167x、171x、174x-179x、181x-184x、189x-196x、198x-214x、219x-229x、231x、232x、236x-239x、243x-245x、248x-256x、258x-276x、279x-282x、285x-288x和290x-295x或其盐以及化合物229H、230、255、256、262和274或其盐。在另一个实施方案中,化合物是式(Vf)化合物,其中R4不是氢,m是0且q是1,且(i)-(iii)中至少一项适用:(i)Q是被取代的或未被取代的芳基、被取代的或未被取代的C3-C8环烷基或被取代的或未被取代的C3-C8环烯基;(ii)R8a、R8b、R8e和R8f中至少一个是羟基、C1-C8烷基或与其所连接的碳以及一个孪位的R8一起形成环烷基部分或羰基部分;和(iii)Q不是哌啶基、氮杂环庚烷基、6-甲基-3-吡啶基或4-吡啶基。在一个实施方案中,化合物是式(Vg)化合物,条件是该化合物不是化合物13x、14x、30x、54x、55x、57x、58x、61x、62x、65x、72x、77x、78x、80x、82x、83x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、132x、135x、138x、140x、141x、142x、143x、144x、145x、146x、157x、160x、161x、163x、166x、170x、177x、179x、181x、182x、183x、187x、190x、192x、195x、197x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、221x、222x、223x、225x、226x、227x、228x、230x、236x、237x、248x、259x、260x、261x、266x、267x和268x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(Vg)化合物,其包括表1中所列的那些化合物,诸如化合物13x、14x、30x、54x、55x、57x、58x、61x、62x、65x、72x、77x、78x、80x、82x、83x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、132x、135x、138x、140x-146x、157x、160x、161x、163x、166x、170x、177x、179x、181x-183x、187x、190x、192x、195x、197x、202x、204x-213x、221x-223x、225x-228x、230x、236x、237x、248x、259x-261x和266x-268x或其盐。在又一个实施方案中,化合物是式(Vg)化合物,其中R4不是氢,条件是该化合物不是14x、80x、82x、83x、132x、141x、170x、187x、197x、216x、217x或230x。在又一个实施方案中,化合物是式(Vg)化合物,其中R4不是氢,m是0且q是1,且Q是未被取代的或被取代的芳基或未被取代的或被取代的杂芳基,而不是4-吡啶基或6-甲基-3-吡啶基。在一个实施方案中,化合物是式(Vh)化合物,条件是该化合物不是化合物13x、30x、54x、55x、57x、58x、61x、62x、65x、71x、72x、77x、78x、88x、89x、92x、93x、94x、96x、97x、100x、101x、104x、105x、125x、126x、127x、128x、129x、130x、135x、138x、140x、142x、143x、144x、145x、146x、157x、160x、161x、163x、166x、177x、179x、181x、182x、183x、185x、190x、192x、195x、202x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、221x、222x、223x、225x、226x、227x、228x、236x、237x、246x、247x、248x、257x、259x、260x、261x、266x、267x、268x、277x、278x、283x、284x和289x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(Vh)化合物,其包括表1中所列的那些化合物,诸如化合物13x、30x、54x、55x、57x、58x、61x、62x、65x、71x、72x、77x、78x、88x、89x、92x-94x、96x、97x、100x、101x、104x、105x、125x-130x、135x、138x、140x、142x-146x、157x、160x、161x、163x、166x、177x、179x、181x-183x、185x、190x、192x、195x、202x、204x-213x、221x-223x、225x-228x、236x、237x、246x-248x、257x、259x-261x、266x-268x、277x、278x、283x、284x和289x或其盐。在另一个实施方案中,化合物是化合物式(Vh),其中R4不是氢,条件是该化合物不是化合物71x、185x、246x、247x、257x、277x、278x、283x、284x和289x中的任何一种。在又一个实施方案中,化合物是式(Vh)化合物,其中R4不是氢,m是0且q是1且Q是被取代的或未被取代的芳基或被取代的杂芳基。在又一个实施方案中,化合物是式(Vh)化合物,其中R4是卤素或烷基,m是0且q是1且Q是被取代的或未被取代的苯基或被取代的吡啶基。在一个实施方案中,化合物是式(Vk)化合物,条件是R1是NRaRb且:(a)Ra和Rb各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基和杂环和被取代的杂环,条件是Ra和Rb基团不都是H;或(b)Ra和Rb与氮原子一起形成杂环或被取代的杂环。在(Vk)的一个实施方案中,其中R1如紧接着的上文所定义,且该化合物进一步具有至少一个以下的结构特征:(i)当m和q是0时,Ra和Rb中至少一个不是甲基,和(ii)当m和q是0时,(a)Q不是被取代的或未被取代的苯基或(b)Q是被取代的或未被取代的杂芳基、被取代的或未被取代的C3-8环烷基、被取代的或未被取代的C3-8环烯基或被取代的或未被取代的杂环基。在式(Vk)的一个实施方案中,m和q中至少一个是1,R1是NRaRb,且(a)Ra和Rb基团各自独立地选自H、烷基、被取代的烷基、链烯基、被取代的链烯基、炔基、被取代的炔基、芳基、被取代的芳基、杂芳基、被取代的杂芳基、杂环和被取代的杂环,条件是Ra和Rb基团不都是H,或(b)Ra和Rb与氮原子一起形成杂环或被取代的杂环。在一个实施方案中,化合物是式(Vn)化合物,条件是(i)该化合物不是化合物1x、2x、3x、4x、5x、6x、7x、8x、9x、10x、11x、12x、13x、14x、15x、16x、17x、18x、19x、20x、21x、22x、23x、24x、25x、26x、27x、28x、29x、30x、31x、32x、33x、34x、35x、36x、37x、38x、39x、40x、41x、42x、43x、44x、47x、49x、50x、52x、54x、55x、56x、57x、58x、59x、60x、61x、62x、65x、72x、77x、78x、80x、81x、82x、83x、84x、85x、88x、89x、90x、92x、93x、94x、95x、96x、97x、98x、100x、101x、102x、103x、104x、105x、106x、107x、109x、110x、111x、113x、115x、116x、117x、118x、119x、120x、121x、122x、123x、124x、125x、126x、127x、128x、129x、130x、131x、132x、134x、135x、136x、137x、138x、139x、140x、141x、142x、143x、144x、145x、146x、148x、149x、150x、151x、152x、155x、157x、158x、159x、160x、161x、162x、163x、164x、165x、166x、167x、168x、169x、170x、171x、173x、174x、175x、176x、177x、178x、179x、181x、182x、183x、184x、186x、187x、189x、190x、191x、192x、193x、194x、195x、196x、197x、198x、199x、200x、201x、202x、203x、204x、205x、206x、207x、208x、209x、210x、211x、212x、213x、214x、219x、220x、221x、222x、223x、224x、225x、226x、227x、228x、229x、230x、231x、232x、236x、237x、238x、239x、243x、244x、245x、248x、249x、250x、251x、252x、253x、254x、255x、256x、258x、259x、260x、261x、262x、263x、264x、265x、266x、267x、268x、269x、270x、271x、272x、273x、274x、275x、276x、279x、280x、281x、282x、285x、286x、287x、288x、290x、291x、292x、293x、294x和295x中的任何一种或其盐且(ii)该化合物不是化合物229H、230、255、256、262和274中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物及施用所述化合物的方法包括任何式(Vn)化合物,其包括表1中所列的那些化合物,诸如化合物1x-44x、47x、49x、50x、52x、54x-62x、65x、72x、77x、78x、80x-85x、88x-90x、92x-98x、100x-107x、109x-111x、113x、115x-132x、134x-146x、148x-152x、155x、157x-171x、173x-179x、181x-184x、186x、187x、189x-214x、219x-232x、236x-239x、243x-245x、248x-256x、258x-276x、279x-282x、285x-288x和290x-295x或其盐以及化合物229H、230、255、256、262和274或其盐。在又一个实施方案中,化合物是式(Vn)化合物,其中R4不是氢,条件是该化合物不是113x、131x、168x、169x、173x或186x。在又一个实施方案中,化合物是式(Vn)化合物,其中R4各自不是氢且至少一个R4是氟或羟基。在又一个实施方案中,化合物是式(Vn)化合物,其中R4各自独立地是卤素、羟基或烷基,条件是(i)至少一个R4是氟或羟基或(ii)各R4基团不相同且Q不是6-甲基-3-吡啶基。在一个实施方案中,化合物是式(Vo)化合物,条件是该化合物不是化合物233x、234x和235x中的任何一种。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(Vzv)化合物,其包括表1中所列的那些化合物,诸如233x、234x和235x。在另一个实施方案中,化合物是式(Vo)化合物,条件是当Q是被取代的哌嗪基时,R8e和R8f不与它们所连接的碳一起形成羰基。在一个实施方案中,化合物是式(Vzv)化合物,条件是该化合物不是化合物45x、51x、216x和217x中的任何一种或其盐。在另一个实施方案中,本发明化合物和本文详述的使用所述化合物的方法包括任何式(Vzv)化合物,其包括表1中所列的那些化合物,诸如45x、51x、216x和217x或其盐。在又一个实施方案中,化合物是式(Vzv)化合物,其中R4不是氢。在又一个实施方案中,化合物是式(Vzv)化合物,其中R4是烷基(例如,甲基)。在一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中任何一个的化合物,其中R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H、羟基、未被取代的C1-C4烷基或与其所连接的碳及一个孪位的R8一起形成环烷基部分。当可适用时,这样的实施方案同样适用于本文详述的任何式的化合物,诸如式(A)-(G)。在一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中R8a、R8b、R8c、R8d、R8e和R8f中至少一个与其所连接的碳以及一个孪位的R8一起形成羰基部分。在另一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H、羟基、甲基或与其所连接的碳及一个孪位的R8一起形成环丙基部分。当可适用时,这样的实施方案同样适用于本文详述的任何式的化合物,诸如式(A)-(G)。在又一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中q是0且m是1。当可适用时,这样的实施方案同样适用于本文详述的任何式的化合物,诸如式(A)-(G)。本发明化合物还包括式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)的中任何一个的化合物,其中q和m都是0。当可适用时,这样的实施方案同样适用于本文详述的任何式的化合物,诸如式(A)-(G)。本发明还包括式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中q、m、R8a、R8b、R8c、R8d、R8e和R8f一起形成选自以下的基团:-CH2-、-CH2CH2-、-CH2CH2CH2-、-CH2-C(H)(OH)-、-C(H)(OH)-CH2-、-CH2-C(OH)(CH3)-、-C(OH)(CH3)-CH2-、-CH2-C(H)(CH3)-、-C(H)(CH3)-CH2-、-CH2-C(CH3)(CH3)-、-C(CH2CH2)-CH2-和-CH2-C(CH2CH2)-。当可适用时,这样的实施方案同样适用于本文详述的任何式的化合物,诸如式(A)-(G)。例如,以式A举例,本发明包括具有以下所列结构的化合物:本发明进一步包括式(F)或本文之前详述的任何实施方案的化合物,其中q、m、R8a、R8b、R8c、R8d、R8e和R8f一起形成选自以下的基团-C(H)(C(O)OH)-CH2-、-C(H)(C(O)OEt)-CH2-、-C(H)(C(O)OMe)-CH2-、-CH2-C(H)(C(O)OH)-、-CH2-C(H)(C(O)OEt)-、-CH2-C(H)(C(O)OMe)-、-C(OH)(CF3)-CH2-和-CH2-C(OH)(CF3)-。在另一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中R4各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、C1-C8全卤代烷基、被取代的或未被取代的杂环基或被取代的或未被取代的芳基。在又一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中R4各自独立地是H或被取代的或未被取代的C1-C8烷基。在又一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中R4各自是H。本发明还包括式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中R4各自独立地是H、卤素、未被取代的C1-C4烷基、C1-C4全卤代烷基或被取代的或未被取代的芳基。本发明进一步包括式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中R4各自独立地是H、卤素、甲基、全氟代甲基或环丙基。本发明还包括式(I)或(Ia)或本文之前详述的任何实施方案的化合物或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基,其可以是但不限于被取代的或未被取代的:吡啶基、苯基、嘧啶基、吡嗪基、咪唑基、呋喃基、吡咯基或噻吩基。在一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是被取代的或未被取代的:苯基或吡啶基。在一个特定的实施方案中,Q是被至少一个甲基所取代的:苯基或吡啶基。在另一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是吡啶基、苯基、嘧啶基、吡嗪基、咪唑基、呋喃基、吡咯基或噻吩基,其被至少一个被取代的或未被取代的C1-C8烷基、卤素或全卤代烷基所取代。在又一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是被取代的或未被取代的C3-8环烷基或被取代的或未被取代的杂环基。在又一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是被取代的或未被取代的:吡啶基、苯基、吡嗪基、哌嗪基、吡咯烷基或硫代吗啉基。在一个特定的实施方案中,Q是吡啶基、苯基、吡嗪基、哌嗪基、吡咯烷基或硫代吗啉基,其被至少一个甲基或卤素所取代。在一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是未被取代的C3-8环烷基或未被取代的杂环基。在另一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是被取代的或未被取代的:环己基、吗啉基、哌嗪基、硫代吗啉基、环戊基或吡咯烷基。在又一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是被取代的:环己基、吗啉基、哌嗪基、硫代吗啉基、环戊基或吡咯烷基,其被至少一个羰基、羟基甲基、甲基或羟基所取代。在又另一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是选自以下结构的基团:其中R9各自独立地是卤素、氰基、硝基、全卤代烷基、全卤代烷氧基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、酰基、酰氧基、羰基烷氧基、烷硫基、被取代的或未被取代的杂环基、烷氧基、被取代的或未被取代的氨基、酰基氨基、磺酰基氨基、磺酰基、羰基、氨基酰基或氨基羰基氨基。在一个实施方案中,Q被不超过一个R9基团所取代。在另一个实施方案中,Q被仅一个R9基团所取代。在一个实施方案中,Q被两个R9基团所取代。在又一个实施方案中,Q选自所详述的芳族结构,其中该基团具有(R9)0,因而Q不含R9官能团或具有式N-R9的部分。在本文详述的包含R9的任何Q基团的另一个实施方案中,应理解本发明包括其中具有超过两个R9基团的实施方案。例如,虽然描述了具有0-2个R9基团的苯基的实施方案,本文仍提供了具有四个R9基团的苯基。在一个实施方案中,当可适用时,Q基团包含3-5个R9基团。在另一个实施方案中,本发明化合物是式(I)、(A)、(E)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是选自以下结构的基团:在又一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是选自以下结构的基团:且其中R9连接于Q的连接与带有R8e和R8f的碳的位置的邻位或对位。在一个特定的实施方案中,Q是下式的结构:且R9连接于的连接与带有R8e和R8f的碳的位置的对位。在另一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是选自以下结构的基团:其中R9各自独立地是卤素、氰基、硝基、全卤代烷基、全卤代烷氧基、被取代的或未被取代的C1-C8烷基、被取代的或未被取代的C2-C8链烯基、被取代的或未被取代的C2-C8炔基、酰基、酰氧基、羰基烷氧基、烷硫基、烷氧基、被取代的或未被取代的氨基、酰基氨基、磺酰基氨基、磺酰基、羰基、氨基酰基或氨基羰基氨基。在一个实施方案中,Q被不超过一个R9基团所取代。在另一个实施方案中,Q被仅一个R9基团所取代。在又一个实施方案中,Q被两个R9基团所取代。在一个特定的实施方案中,Q选自所详述碳环和杂环结构,其中该基团具有(R9)0,因而Q不含R9官能团或具有式N-R9的部分。在本文详述的任何结构或实施方案中,在一个实施方案中,Q是:在本文详述的包含R基团的任何结构或实施方案中,在一个实施方案中,R9各自独立地是被取代的或未被取代的C1-C4烷基、卤素、三氟甲基或羟基。在另一个实施方案中,R9各自独立地是甲基、-CH2OH、异丙基、卤素、三氟甲基或羟基。在另一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是选自以下结构的基团:在另一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是选自以下结构的基团:在又一个实施方案中,本发明化合物是式(I)或(Ia)或本文之前详述的任何实施方案的化合物,或是式(IIa)-(IIh)、(IIIa)-(IIIm)、(IVa)-(IVk)或(Va)-(Vzv)中的任何一个的化合物,其中Q是选自以下结构的基团:在又一个实施方案中,本发明化合物是式(I)化合物,其中R1是未被取代的烷基,R2a、R2b、R3a、R3b、R10a和R10b各自是H,X7、X8、X9和X10各自独立地是N或CH,R8a、R8b、R8c、R8d、R8e和R8f各自独立地是H或羟基,且Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基,其包括但不限于被取代的或未被取代的:苯基或吡啶基。当Q是被取代的:苯基或吡啶基,在一个实施方案中,其是被至少一个甲基所取代。在又一个实施方案中,本发明化合物是式(I)化合物,其中R1是被取代的或未被取代的C1-C8烷基、酰基、酰氧基、羰基烷氧基、被取代的或未被取代的杂环基、被取代的或未被取代的芳基;R2a和R2b各自独立地是H、未被取代的C1-C8烷基或卤素;R3a和R3b各自独立地是H或卤素;X7、X8、X9和X10各自是CR4,其中R4是如在式(I)中所定义,或在一个特定的实施方案中,R4是H、卤素、吡啶基、甲基或三氟甲基;R10a和R10b都是H,且Q是被取代的或未被取代的芳基、被取代的或未被取代的杂芳基,其包括但不限于被取代的或未被取代的:吡啶基、苯基、嘧啶基、吡嗪基、咪唑基、呋喃基、吡咯基或噻吩基。在一个特定的实施方案中,Q是吡啶基、苯基、嘧啶基、吡嗪基、咪唑基、呋喃基、吡咯基或噻吩基,其被至少一个被取代的或未被取代的C1-C8烷基、卤素或全卤代烷基所取代。在一个实施方案中,提供了本文详述的实施方案的化合物,其中R1是丙醇基(propylate)、甲基、乙基、环丙基、三氟甲基、异丙基、叔-丁基、仲-丁基、2-甲基丁基、丙醛、1-甲基-2-羟基乙基、2-羟基乙醛、2-羟基乙基、2-羟基丙基、2-羟基-2-甲基丙基、环丁基、环戊基、环己基、被取代的苯基、哌啶-4-基、羟基环戊-3-基、羟基环戊-2-基、羟基环丙-2-基、1-羟基-1-甲基环丙-2-基或1-羟基-1,2,2-三甲基-环丙-3-基。在又一个实施方案中,本发明化合物是式(I)化合物,其中R1是被取代的或未被取代的C1-C8烷基;R2a、R2b、R3a和R3b各自独立地是H或卤素;X1是N;R4各自独立地是H、卤素、C1-C8全卤代烷基、被取代的或未被取代的C1-C8烷基;R8a、R8b、R8c、R8d、R8e和R8f各自是H;且Q是被取代的或未被取代的:环己基、吗啉基、哌嗪基、硫代吗啉基、环戊基或吡咯烷基。本发明还包括式(I)化合物,其中R1是甲基;X7、X8、X9和X10中至少一个是CR4,且R4各自独立地是H、卤素、甲基或三氟甲基。本发明包括其中在所详述的任何实施方案中的Q被至少一个羰基、羟基甲基、甲基或羟基所取代的化合物。在一个特定的实施方案中,化合物是式(I)化合物,其中R1是被取代的或未被取代的C1-C8烷基;R2a和R2b各自独立地是H、被取代的或未被取代的C1-C8烷基或R2a和R2b一起形成羰基部分;R3a和R3b都是H;X1是N,R4各自独立地是H、卤素或被取代的或未被取代的C1-C8烷基;R8a、R8b、R8c、R8d、R8e和R8f各自是H;R10a和R10b各自独立地是H、卤素、被取代的或未被取代的C1-C8烷基、羟基、烷氧基或R10a和R10b一起形成羰基,条件是R10a和R10b中至少一个不是H。在该实施方案的一个方面,Q可以是被取代的或未被取代的:吡啶基、苯基、吡嗪基、哌嗪基、吡咯烷基或硫代吗啉基。在该实施方案的另一个方面,Q是吡啶基、苯基、吡嗪基、哌嗪基、吡咯烷基或硫代吗啉基,其被至少一个甲基或卤素所取代。在该实施方案的又一个方面,X7、X8、X9和X10是CR4且R4各自独立地是H、卤素或甲基。在一个特定的实施方案中,本发明包括式(F)化合物或其盐或溶剂化物:其中:R4是卤素或C1-C4烷基;Q是R9是卤素、烷氧基或H,条件是当R9是H时,R4不是氟或甲基;且R9A是全卤代烷基或烷基,条件是R9A不是CH3。在式(F)的一个实施方案中Q是在一个此类实施方案中,R4是氯。在又一个实施方案中,R4是氯,且R9是氢。在的另一个式(F)的实施方案中,Q是在一个此类实施方案中,R4是卤素。在另一个此类实施方案中,R9A是全卤代烷基。在又一个此类实施方案中,R4是卤素,且R9A是全卤代烷基(例如,当R4是氯且R9A是三氟甲基时)。在又一个此类实施方案中,R9A是C1-C4烷基(例如,丙基,其在一个实施方案中是正-丙基且在另一个中是异丙基)。在又另一个式(F)的实施方案中,Q是本发明化合物的实例在表2中描述。即使没有描述盐,所述化合物也可以以盐形式出现,且应理解本发明包括本文所述化合物的所有盐和溶剂化物,以及所述化合物的非盐和非溶剂化物形式,这也是技术人员所公知的。表2.本发明代表性的化合物。本发明另外的化合物包括式(G)化合物或其可药用盐:其中:R1选自未被取代的烷基和未被取代的芳烷基或杂芳烷基;R19选自氢、未被取代的烷基、未被取代的烷氧基、和卤素;且Q选自被取代的噻唑基、被取代的三唑基和被取代的二唑基。在一个实施方案中,Q是三唑-硫酮(triazolo-thione)。在式(G)的一些实施方案或本文的任何实施方案中,诸如式(Ga)-(Gd)化合物中,R19选自氢、未被取代的C1-C8烷基、未被取代的C1-C8烷氧基和卤素。在一些实施方案中,R19选自氢、未被取代的C1-C4烷基、未被取代的C1-C4烷氧基和卤素。在一些实施方案中,R19选自氢、甲基、甲氧基、氟和氯。在另一个实施方案中,本发明化合物是式G化合物或本文的任何实施方案,诸如式(Ga)-(Gd)化合物,其中A-环选自以下结构:在式(G)的一些实施方案或其本文的任何实施方案中,诸如式(Ga)-(Gd)化合物中,其中R1选自未被取代的C1-C8烷基和未被取代的C1-C8芳烷基或杂芳烷基。在一些实施方案中,R1选自未被取代的C1-C4烷基和未被取代的C1-C4芳烷基或杂芳烷基。在一些实施方案中,R1选自甲基和苄基。还应理解本文详述的任何R1可与R19的任何实施方案组合。在一个实施方案中,R1和R19都是甲基。在又一个实施方案中,本发明化合物是式G化合物或其本文的任何实施方案,诸如式(Ga)-(Gd)化合物,其中C-环选自以下结构:在一些实施方案中,Q选自噻唑-2-基、1,2,4-三唑-3-基、5-硫代-4,5-二氢-1H-1,2,4-三唑-3-基和1,2,4-二唑-3-基。在一些实施方案中,Q是噻唑-2-基、1,2,4-三唑-3-基、5-硫代-4,5-二氢-1H-1,2,4-三唑-3-基或1,2,4-二唑-3-基,其被选自未被取代的C1-C8烷基和未被取代的芳基或杂芳基的基团所取代。在一些实施方案中,Q是噻唑-2-基、1,2,4-三唑-3-基、5-硫代-4,5-二氢-1H-1,2,4-三唑-3-基或1,2,4-二唑-3-基,其被选自未被取代的C1-C4烷基和未被取代的芳基或杂芳基的基团所取代。在一些实施方案中,Q是噻唑-2-基、1,2,4-三唑-3-基、5-硫代-4,5-二氢-1H-1,2,4-三唑-3-基或1,2,4-二唑-3-基,其被选自甲基、苯基和吡啶-4-基的基团所取代。在另一个实施方案中,本发明化合物是式G化合物,其中环A和C如本文的任何实施方案中所详述,且Q选自以下结构:其中R13a选自氢和烷基;R14a选自芳基和杂芳基;R13b是烷基;R13c选自烷基、芳基和杂芳基;且R13d选自芳基和杂芳基。在另一个实施方案中,Q选自以上结构,且R13a选自氢和甲基;R14a选自苯基和吡啶-4-基;R13b是甲基;R13c选自甲基、苯基和吡啶-4-基;且R13d选自苯基和吡啶-4-基。因而,本发明包括式(Ga)-(Gd)化合物:在一些实施方案中,化合物是式Ga化合物,其中R1选自甲基和苄基;R19选自氢、甲基、甲氧基、氯和氟;R13a选自氢和甲基;且R14a选自苯基和吡啶-4-基。在一些实施方案中,化合物是式Gb化合物,其中R1选自甲基和苄基;R19选自氢、甲基、甲氧基、氯和氟;且R13b是甲基。在一些实施方案中,化合物是式Gc化合物,其中R1选自甲基和苄基;R19选自氢、甲基、甲氧基、氯和氟;且R13c选自甲基、苯基和吡啶-4-基。在一些实施方案中,化合物是式Gd化合物,其中R1选自甲基和苄基;R19选自氢、甲基、甲氧基、氯和氟;且R13d选自苯基和吡啶-4-基。本发明化合物的某些实例在表3中描述。即使没有描述盐,所述化合物也可以以盐形式存在,且应理解本发明包括本文所述化合物的所有盐、水合物和溶剂化物,以及所述化合物的非盐、非水合物和非溶剂化物形式,这也是技术人员所公知的。因而理解到的是本发明还涉及本发明化合物的可药用盐。表3.本发明代表性的化合物如表3中所列的本发明化合物在下文生物实施例16B至19B中还被称为:G6-a(C4-1);G6-b(C4-5);G6-c(C4-4);G6-d(C4-6);G6-e(C4-3);G6-f(C4-7);G8-a(C1-1);G8-b(C1-6);G8-c(C1-5);G8-d(C1-4);G8-e(C1-8);G8-f(C1-7);G11-a(C2-1);G11-b(C2-5);G11-c(C2-6);和G11-d(C2-4)。包含本文详述的任何化合物的药物组合物包括在本发明内。因而,本发明包括含有本发明化合物或其可药用盐及可药用载体或赋形剂的药物组合物。本发明提供了包含本文所公开的化合物或其可药用盐(例如,草酸盐或TFA盐或盐酸盐或二盐酸盐)及可药用载体或赋形剂的药物组合物。本发明的药物组合物可以采取适用于口服、经颊、胃肠外、经鼻、局部或直肠给药的形式,或适于通过吸入给药的形式。在一个实施方案中,本发明化合物是为施用于个体而制备的合成的化合物。在另一个实施方案中,提供了包含以基本上纯的形式存在的本发明化合物的组合物。在另一个实施方案中,本发明包括含有本发明化合物和可药用载体的药物组合物。在另一个实施方案中,提供了施用本发明化合物的方法。纯化的形式、药物组合物和施用所述化合物的方法适用于本文详述的任何化合物或其形式,诸如化合物229H、230、241、255、256、262和274。特别地,当适用时,在一个实施方案中Dimebon的代谢产物被排除在所提供的通式的化合物之外。在另一个实施方案中,本发明化合物的方法、药物组合物、分离的和纯化的形式包括Dimebon的代谢产物。认为所述代谢产物包括化合物229H、230、241、255、256、262、274和实施例93的化合物。在一个实施方案中,被一个O-Glu取代的化合物同样被排除在所提供的通式的化合物之外。在一个实施方案中,本发明化合物依据任何本文所详述的式,其中化合物进一步是本发明的1型化合物。在一个实施方案中,本发明化合物依据任何本文所详述的式,其中化合物进一步是本发明的2型化合物。在一个实施方案中,本发明化合物依据任何本文所详述的式,其中化合物进一步是本发明的3型化合物。在一个实施方案中,本发明化合物依据任何本文所详述的式,其中化合物进一步是本发明的4型化合物。化合物分类可以测定本文公开的化合物与一组胺能G蛋白偶联受体的结合特性,所述胺能G蛋白偶联受体包括肾上腺素能受体、多巴胺受体、血清素受体、组胺受体和咪唑啉受体。结合特性可通过本领域已知的方法评价,诸如竞争性结合试验。在一个实施方案中,化合物通过本文详述的结合试验评价。还可在基于细胞的试验或在体内模型中测定本文公开的化合物,来进一步表征。在一方面,本文公开的化合物是本文详述的任何式的化合物,且还显示其一种或多种以下的特性:对配体结合于肾上腺素能受体(例如,α1D、α2A和α2B)的抑制、对配体结合于血清素受体(例如,5-HT2A、5-HT2C、5-HT6和5-HT7)的抑制、对配体结合于多巴胺受体(例如,D2L)的抑制,和对配体结合于组胺受体(例如,H1、H2和H3)的抑制;对血清素受体(例如,5-HT2A、5-HT6)的激动剂/拮抗剂活性;对多巴胺受体(例如,D2L、D2S)的激动剂/拮抗剂活性;对组胺受体(例如,H1)的激动剂/拮抗剂活性;在神经突向外生长试验中的活性;在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效;和在精神分裂症的临床前模型中的功效。在一个实施方案中,在本文所述的试验中测定对配体结合于受体的抑制。在另一个实施方案中,在本领域已知的试验中测定对配体结合于受体的抑制。在一个实施方案中,配体与受体的结合被抑制至少约80%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,配体与受体的结合被抑制超过约80%、85%、90%、95%、100%中的任何一个或约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,配体与受体的结合被抑制至少约80%±20%,如在本领域已知的试验中所测定的那样。在一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个本文详述的受体(例如,a1D、a2A、a2B、5-HT2A、5-HT2C、5-HT6、5-HT7、D2L、H1、H2、H3),且还显示其了对一种或多种本文详述的受体(例如,5-HT2A、5-HT6、D2L、D2S、H1)的激动剂或拮抗剂活性。在一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,且还刺激神经突向外生长,例如,如通过本文所述的试验中所测定的那样。在另一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,还显示其对一种或多种本文详述的受体的激动剂或拮抗剂活性,且还刺激神经突向外生长。在又一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个本文详述的受体,且还显示其在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效。在另一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个本文详述的受体,还显示其在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效,且还显示其对一种或多种本文详述的受体的激动剂或拮抗剂活性。在又一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个本文详述的受体,还显示其在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效,并且还刺激神经突向外生长。在另一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个本文详述的受体,还其显示在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效,还显示其对一种或多种本文详述的受体的激动剂或拮抗剂活性,并且还刺激神经突向外生长。在又一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,且还显示其在精神分裂症的临床前模型中的功效。在另一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,还显示其在精神分裂症的临床前模型中的功效,且还显示其对一种或多种本文详述的受体的激动剂或拮抗剂活性。在又一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,还显示其在精神分裂症的临床前模型中的功效,并且还刺激神经突向外生长。在又一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,还其显示在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效,且还显示其在精神分裂症的临床前模型中的功效。在另一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,还显示其在精神分裂症的临床前模型中的功效,还显示其对一种或多种本文详述的受体的激动剂或拮抗剂活性,且还显示其在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效。在另一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,还显示其在精神分裂症的临床前模型中的功效,还刺激神经突向外生长,且还显示其在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效。在又一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个本文详述的受体,还显示其对一种或多种本文详述的受体的激动剂或拮抗剂活性,还刺激神经突向外生长,且还显示其在精神分裂症的临床前模型中的功效。在另一个实施方案中,本发明化合物抑制配体结合于至少一个和多达11个如本文详述的受体,还显示其在精神分裂症的临床前模型中的功效,还显示其对一种或多种本文详述的受体的激动剂或拮抗剂活性,还刺激神经突向外生长,且还显示其在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效。在一方面,本发明化合物抑制配体结合于肾上腺素能受体α1D、α2A、α2B,并抑制配体结合于血清素受体5-HT6。在另一个实施方案中,本发明化合物抑制配体结合于肾上腺素能受体α1D、α2A、α2B,抑制配体结合于血清素受体5-HT6,并抑制配体结合于以下受体中的一种或多种:血清素受体5-HT7、5-HT2A和5-HT2C。在另一个实施方案中,本发明化合物抑制配体结合于肾上腺素能受体α1D、α2A、α2B,抑制配体结合于血清素受体5-HT6,并抑制配体结合于以下受体中的一种或多种:血清素受体5-HT7、5-HT2A和5-HT2C,且还显示微弱地抑制配体结合于组胺受体H1和/或H2。在另一个实施方案中,本发明化合物抑制配体结合于肾上腺素能受体α1D、α2A、α2B,抑制配体结合于血清素受体5-HT6,且还显示微弱地抑制配体结合于组胺受体H1和/或H2。在另一个实施方案中,本发明化合物抑制配体结合于多巴胺受体D2L。在另一个实施方案中,本发明化合物抑制配体结合于多巴胺受体D2L,并抑制配体结合于血清素受体5-HT2A。在另一个实施方案中,本发明化合物抑制配体结合于组胺受体H1。在一个实施方案中,结合被抑制至少约80%,如在适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,配体与受体的结合被抑制超过约80%、85%、90%、95%、100%中的任何一个或约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,本发明化合物显示本文详述的任何的受体结合特性,且还显示其对以下受体中的一种或多种的激动剂/拮抗剂活性:血清素受体5-HT2A、血清素受体5-HT6、多巴胺受体D2L、多巴胺受体D2S和组胺受体H1。在一个实施方案中,本发明化合物显示本文详述的任何的受体结合特性,并且还刺激神经突向外生长。在一个实施方案中,本发明化合物显示本文详述的任何的受体结合特性,且还显示其在与胆碱能功能亢进相关的记忆功能障碍的临床前模型中的功效。在一个实施方案中,本发明化合物显示本文详述的任何的受体结合特性,且还显示其在精神分裂症的临床前模型中的功效。在一个实施方案中,本发明化合物显示本文详述的任何的受体结合特性,且还显示其在任何的一种或多种激动剂/拮抗剂试验(例如,对血清素受体5-HT2A、5-HT6、多巴胺受体D2L、多巴胺受体D2S和组胺受体H1的激动剂/拮抗剂功效)、神经突向外生长、胆碱能功能亢进相关的记忆功能障碍的临床前模型和精神分裂症的临床前模型中的功效。抑制配体结合于肾上腺素能受体a1D、a2A、a2B和血清素受体5-HT6至少约80%(如在适合的试验诸如本文所述的试验中所测定的)的本发明化合物可以被归类为1型化合物。在一个实施方案中,配体与肾上腺素能受体a1D、a2A、a2B和血清素受体5-HT6的结合被抑制超过约80%、85%、90%、95%、100%中的任何一个、约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。特别需要还显示出对配体结合于血清素受体5-HT7有强的抑制作用的1型化合物。在一个实施方案中,配体与血清素受体5-HT7的结合被抑制至少约80%。在另一个实施方案中,配体与血清素受体5-HT7的结合被抑制超过约80%、85%、90%、95%、100%中的任何一个、约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。允许微弱抑制配体与组胺H1受体的结合,原因是已知该受体的激动剂牵涉于刺激记忆以及体重增加。在一个实施方案中,与组胺受体H1的结合被抑制小于约80%。在另一个实施方案中,配体与组胺受体H1的结合被抑制小于约以下各值中的任何一个:75%、70%、65%、60%、55%或50%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。抑制配体结合于多巴胺D2L受体至少约80%(如在适合的试验诸如本文所述的试验中所测定的)的化合物可以被归类为2型化合物。不抑制配体结合于肾上腺素能受体a1D、a2A、a2B、血清素受体5-HT6和多巴胺受体D2L至少约80%(如在适合的试验诸如本文所述的试验中所测定的)的、但在神经突向外生长试验中显示活性的本发明化合物可以被归类为3型化合物。不抑制配体结合于肾上腺素能受体a1D、a2A、a2B、血清素受体5-HT6和多巴胺受体D2L至少约80%(如在适合的试验诸如本文所述的试验中所测定的)的、且在神经突向外生长试验中不显示活性的或如果神经突向外生长所需其的浓度超过1μM的本发明化合物被称为4型化合物。1型化合物1型化合物抑制配体结合于肾上腺素能受体a1D、a2A、a2B和血清素受体5-HT6至少约80%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,1型化合物分别抑制哌唑嗪、MK-912或萝芙辛结合于肾上腺素能受体a1D、a2A和a2B并抑制LSD结合于血清素受体5-HT6至少约80%,如在本文所述的试验中所测定的那样。在另一个实施方案中,结合被抑制超过约80%、85%、90%、95%、100%中的任何一个、约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在另一个实施方案中,抑制配体结合至少约80%±20%,如通过本文领域技术人员已知的试验所测定的那样。在一个实施方案中,1型化合物还抑制配体结合于多巴胺受体D2L小于约80%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在某些方面,1型化合物还显示一种或多种以下的特性:强烈抑制配体结合于血清素5-HT7受体、强烈抑制配体结合于血清素5-HT2A受体、强烈抑制配体结合于血清素5-HT2C受体、微弱抑制配体结合于组胺H1受体、微弱抑制配体结合于组胺H2受体和对血清素受体5-HT2A的拮抗剂活性。在一个实施方案中,配体与血清素受体5-HT7、5-HT2A和/或5-HT2C的结合被抑制至少约80%,如在适合的试验诸如本文所述的试验中所测定的那样。在另一个实施方案中,配体与血清素受体5-HT7、5-HT2A和/或5-HT2C的结合被抑制超过约80%、85%、90%、95%、100%中的任何一个、约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,配体与组胺受体H1和/或H2的结合被抑制小于约80%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在另一个实施方案中,配体与组胺受体H1和/或H2的结合被抑制小于约以下各值中的任何一个:75%、70%、65%、60%、55%或50%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,1型化合物抑制血清素受体5-HT2A的激动剂响应至少约以下各值中的任何一个:50%、60%、70%、80%、90%、100%、110%、120%、130%、140%、150%,如在适合的试验诸如本文所述的试验中所测定的那样。在一些方面,1型化合物显示以上所述的神经递质受体结合特性,且还显示其对一种或多种以下的受体的激动剂或拮抗剂活性:血清素受体5-HT2A、血清素受体5-HT6、多巴胺受体D2L和多巴胺受体D2S、组胺受体H1,如在本文所述的试验所测定的那样。在一个实施方案中,1型化合物抑制血清素受体5-HT2A的激动剂响应至少约以下各值中的任何一个:50%、60%、70%、80%、90%、100%、110%、120%、130%、140%、150%,如在适合的试验诸如本文所述的试验中所测定的那样。在一些方面,1型化合物显示以上所述的神经递质受体结合特性,并且还刺激神经突向外生长,如在适合的试验诸如本文所述的试验中所测定的那样。某些1型化合物在使用原代培养神经元的神经突向外生长试验中显示活性(参见实施例11B)。得到的数据显示本发明化合物具有与自然出现的原型的神经营养蛋白诸如脑源性神经营养因子(BDNF)和神经生长因子(NGF)强度相当的活性。值得注意地是,神经突向外生长起到新的突触发生的关键作用,这对神经元病症的治疗是有益的。在一个实施方案中,在约1μM的效价强度观察到神经突向外生长,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在另一个实施方案中,观察到神经突向外生长,效价强度为约500nM。在又一个实施方案中,观察到神经突向外生长,效价强度为约50nM。在另一个实施方案中,观察到神经突向外生长,效价强度为约5nM。在某些方面,1型化合物显示以上所述的神经递质受体结合特性,且还显示其在记忆功能障碍的临床前模型中的促-认知作用。已证明1型化合物在与胆碱功能减退相关的记忆功能障碍的临床前模型中有效(参见实施例12B)。由于H1拮抗作用可引起镇静、体重增加和认知降低,对该受体的低亲和力(在本文所述的试验中,在1μM抑制美吡拉敏的结合小于约80%)可与促-认知作用和更加需要的副作用特性相关。此外,具有增强的作为5-HT拮抗剂的效力的1型化合物可具有认知增强作用,因为血清素通过该受体所起的作用可损害记忆。在另一个方面,1型化合物显示以上所述的神经递质受体结合特性,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在另一个实施方案中,1型化合物显示以上所述的神经递质受体结合特性,且不具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在一个实施方案中,1型化合物显示以上所述的神经递质结合特性,对至少一种本文详述的受体显示激动剂或拮抗剂活性,并且还刺激神经突向外生长。在又一个实施方案中,1型化合物显示以上所述的神经递质结合特性,还对至少一种本文详述的受体显示激动剂或拮抗剂活性,且还显示其在记忆功能障碍的临床前模型中的促-认知作用。在又一个实施方案中,1型化合物显示以上所述的神经递质结合特性,还对至少一种本文详述的受体显示激动剂或拮抗剂活性,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在又一个实施方案中,1型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,且还显示其在记忆功能障碍的临床前模型中的促-认知作用。在又一个实施方案中,1型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在又一个实施方案中,1型化合物显示以上所述的神经递质结合特性,还显示其在记忆功能障碍的临床前模型中的促-认知作用,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在一个实施方案中,1型化合物显示以上所述的神经递质结合特性,还对至少一种本文详述的受体显示激动剂或拮抗剂活性,还刺激神经突向外生长,且还显示其在记忆功能障碍的临床前模型中的促-认知作用。在一个实施方案中,1型化合物显示以上所述的神经递质结合特性,还对至少一种本文详述的受体显示激动剂或拮抗剂活性,还刺激神经突向外生长,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在一个实施方案中,1型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,还显示其在记忆功能障碍的临床前模型中的促-认知作用,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在一个实施方案中,1型化合物显示以上所述的神经递质结合特性,对至少一种本文详述的受体显示激动剂或拮抗剂活性,还刺激神经突向外生长,还显示其在记忆功能障碍的临床前模型中的促-认知作用,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。因此,一方面,1型化合物对治疗认知和记忆相关障碍特别有用,所述障碍包括但不限于:阿尔茨海默病、亨廷顿病、帕金森病、肌萎缩侧索硬化(ALS)、孤独症、轻度认知功能损害(MCI)、与精神分裂症或其它的精神病性精神障碍相关的认知功能损害、中风、创伤性脑损伤(TBI)和年龄相关的记忆损害(AAMI)和病原体-诱导的认知功能障碍,例如,HIV相关的认知功能障碍、莱姆病相关的认知功能障碍。在另一个方面,1型化合物对治疗神经递质-介导的障碍特别有用,其包括但不限于脊髓损伤、糖尿病性神经病和牵涉于老化保护活性的疾病,诸如年龄相关的掉发(脱发)、年龄相关的体重减轻和年龄相关的视觉障碍(白内障)。本发明1型化合物包括化合物37、26、28、29、32、43、47、45、83、162、89、250、68、357、55、60、404、371、452、56、333、334、360、372、443、444、456、476、487和225。2型化合物2型化合物抑制配体结合于多巴胺受体D2L至少约80%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,2型化合物抑制螺哌隆结合于多巴胺受体D2L至少约80%,如在本文所述的试验中所测定的那样。在另一个实施方案中,抑制结合超过约80%、85%、90%、95%、100%中的任何一个、约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在另一个实施方案中,抑制配体结合至少约80%±20%,如通过本领域技术人员已知的试验所测定的那样。在某些方面,2型化合物还显示对配体结合于血清素受体5-HT2A的强烈抑制,如在本文所述的试验中所测定的那样。在一个实施方案中,对血清素受体5-HT2A的结合被抑制至少约80%,在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在另一个实施方案中,抑制结合超过约80%、85%、90%、95%、100%中的任何一个、约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一些方面,2型化合物抑制配体结合于肾上腺素能受体a1D、a2A、a2B、血清素受体5-HT6和多巴胺受体D2L至少约80%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,2型化合物抑制配体结合于肾上腺素能受体a1D、a2A、a2B、血清素受体5-HT6和多巴胺受体D2L至少约80%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。在另一个实施方案中,2型化合物抑制配体结合于肾上腺素能受体a1D、a2A、a2B、血清素受体5-HT6和多巴胺受体D2L超过约80%、85%、90%、95%、100%中的任何一个、约85-95%或约90-100%,如在本领域已知的适合的试验诸如本文所述的试验中所测定的那样。由于H1拮抗作用可引起镇静、体重增加和认知降低,对该受体的低亲和力(在本文所述的试验中,在1μM抑制美吡拉敏的结合小于约80%)可与促-认知作用和更加需要的副作用特性相关。在一些方面,2型化合物显示以上所述的神经递质受体结合特性,并且还显示其对一种或多种以下的受体的激动剂或拮抗剂活性:血清素受体5-HT2A、血清素受体5-HT6、多巴胺受体D2L、多巴胺受体D2S和组胺受体H1,如在适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,2型化合物抑制对血清素受体5-HT2A的激动剂响应至少约以下各值中的任何一个:50%、60%、70%、80%、90%、100%、110%、120%、130%、140%、150%,如在适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,2型化合物抑制对多巴胺受体D2L的激动剂响应至少约以下各值中的任何一个:50%、60%、70%、80%、90%、100%、110%、120%、130%、140%、150%,如在适合的试验诸如本文所述的试验中所测定的那样。在一个实施方案中,2型化合物抑制对多巴胺受体D2S的激动剂响应至少约以下各值中的任何一个:50%、60%、70%、80%、90%、100%、110%、120%、130%、140%、150%,如在适合的试验诸如本文所述的试验中所测定的那样。在一些方面,2型化合物显示以上所述的神经递质受体结合特性,并且还显示其抗精神病效应。确认了2型化合物具有与具有抗精神病活性的化合物相似的结合特性,且已显示若干2型化合物在精神分裂症的临床前模型中有效(参见实施例13B)。此外,2型化合物可具有Dimebon的认知增强特性,且因而增加这些抗精神病的分子的有益的药理学特性。在一个实施方案中,2型化合物显示以上所述的神经递质受体结合特性,并且还显示其在记忆功能障碍的临床前模型中的促-认知作用。在另一个实施方案中,2型化合物显示以上所述的神经递质受体结合特性,且未显示在记忆功能障碍的临床前模型中有促-认知作用。在一些方面,2型化合物显示以上所述的神经递质受体结合特性,并且还刺激神经突向外生长,如在适合的试验诸如本文所述的试验中所测定的那样。某些2型化合物在使用原代培养神经元的神经突向外生长试验中显示活性(参见实施例11B)。在一个实施方案中,观察到神经突向外生长,效价强度为约1μM。在另一个实施方案中,观察到神经突向外生长,效价强度为约500nM。在又一个实施方案中,观察到神经突向外生长,效价强度为约50nM。在另一个实施方案中,观察到神经突向外生长,效价强度为约5nM。在一个实施方案中,2型化合物显示以上所述的神经递质结合特性,对至少一种本文详述的受体显示激动剂或拮抗剂活性,并且还刺激神经突向外生长。在又一个实施方案中,2型化合物显示以上所述的神经递质结合特性,还对至少一种本文详述的受体显示激动剂或拮抗剂活性,并且还显示其在记忆功能障碍的临床前模型中的促-认知作用。在又一个实施方案中,2型化合物显示以上所述的神经递质结合特性,还对至少一种本文详述的受体显示激动剂或拮抗剂活性,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在又一个实施方案中,2型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,并且还显示其在记忆功能障碍的临床前模型中的促-认知作用。在又一个实施方案中,2型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在又一个实施方案中,2型化合物显示以上所述的神经递质结合特性,还显示其在记忆功能障碍的临床前模型中的促-认知作用,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在一个实施方案中,2型化合物显示以上所述的神经递质结合特性,还对至少一种本文详述的受体显示激动剂或拮抗剂活性,还刺激神经突向外生长,并且还显示其在记忆功能障碍的临床前模型中的促-认知作用。在一个实施方案中,2型化合物显示以上所述的神经递质结合特性,还对至少一种本文详述的受体显示激动剂或拮抗剂活性,还刺激神经突向外生长,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在一个实施方案中,2型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,还显示其在记忆功能障碍的临床前模型中的促-认知作用,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在一个实施方案中,2型化合物显示以上所述的神经递质结合特性,对至少一种本文详述的受体显示激动剂或拮抗剂活性,还刺激神经突向外生长,还显示其在记忆功能障碍的临床前模型中的促-认知作用,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。因此,一方面,2型化合物对治疗精神病性适应症诸如精神分裂症、双相性精神障碍、精神病、抑郁和焦虑特别有用。2型化合物包括化合物25、20、34、32、33、36、247、253、336、349、337和455。3型化合物3型化合物不抑制配体结合于肾上腺素能受体a1D、a2A、a2B;血清素受体5-HT6和多巴胺受体D2L至少约80%,如在适合的试验诸如本文所述的试验中所测定的,但在神经突向外生长试验例如在本文所述的试验中有活性。某些3型化合物在使用原代培养神经元的神经突向外生长试验中显示活性(参见实施例11B)。在一个实施方案中,观察到神经突向外生长,效价强度为小于或等于约1μM。在另一个实施方案中,观察到神经突向外生长,效价强度为约500nM。在又一个实施方案中,观察到神经突向外生长,效价强度为约50nM。在另一个实施方案中,观察到神经突向外生长,效价强度为约5nM。在一些方面,3型化合物显示以上所述的神经递质受体结合特性,并且还显示促-认知作用。由于H1拮抗作用可引起镇静、体重增加和认知降低,对该受体的低亲和力(在本文所述的试验中,在1μM抑制美吡拉敏的结合小于约80%)可与促-认知作用和更加需要的副作用特性相关。在又一个实施方案中,3型化合物显示以上所述的神经递质受体结合特性,并且还具有抗精神病效应。在另一个实施方案中,3型化合物显示以上所述的神经递质受体结合特性,并不具有抗精神病效应。在一个实施方案中,3型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,并且还显示其在记忆功能障碍的临床前模型中的促-认知作用。在另一个实施方案中,3型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。在又一个实施方案中,3型化合物显示以上所述的神经递质结合特性,还刺激神经突向外生长,还显示其在记忆功能障碍的临床前模型中的促-认知作用,并且还具有抗精神病效应,如在精神分裂症的临床前模型中所测定的那样。因此,一方面,3型化合物对治疗神经元病症特别有用,所述神经元病症诸如阿尔茨海默病、亨廷顿病、肌萎缩侧索硬化(ALS)、帕金森病、犬认知功能障碍综合征(CCDS)、雷维小体病、门克斯病、威尔逊病、克雅氏病、法尔病、与脑循环有关的急性或慢性病症诸如缺血性或出血性中风或其它的大脑出血性损伤、年龄相关的记忆损害(AAMI)、轻度认知功能损害(MCI)、与创伤有关的轻度认知功能损害(MCI)、脑震荡后综合征、创伤后应激障碍和辅助化学疗法、创伤性脑损伤(TBI)、神经元死亡介导的眼部疾病、黄斑变性、年龄有关的黄斑变性、孤独症(包括孤独症谱系障碍)、阿斯珀格综合征和雷特综合征、撕脱损伤、脊髓损伤、重症肌无力、吉兰巴雷综合征、多发性硬化、糖尿病性神经病、纤维肌痛、与脊髓损伤相关的神经病、精神分裂症、双相性精神障碍、精神病、焦虑和抑郁。在另一个方面,3型化合物对治疗神经递质-介导的障碍特别有用,其包括但不限于脊髓损伤、糖尿病性神经病、变应性疾病和牵涉于老化保护活性的疾病,诸如年龄相关的掉发(脱发)、年龄相关的体重减轻和年龄相关的视觉障碍(白内障)。3型化合物包括化合物395。4型化合物4型化合物不抑制配体结合于肾上腺素能受体a1D、a2A、a2B、血清素受体5-HT6以及多巴胺受体D2L至少约80%,如在适合的试验诸如本文所述的试验中所测定的,并且在神经突向外生长试验中有效,其效价强度仅是大于约1μM。在一个实施方案中,4型化合物在神经突向外生长试验没有活性。在一个实施方案中,4型化合物在记忆功能障碍的临床前模型中显示促-认知作用。在另一个实施方案中,4型化合物在记忆功能障碍的临床前模型中不显示促-认知作用。在又一个实施方案中,4型化合物在精神分裂症的临床前模型中具有抗精神病效应。在另一个实施方案中,4型化合物在精神分裂症的临床前模型中不具有抗精神病效应。在又一个实施方案中,4型化合物在记忆功能障碍的临床前模型中的显示促-认知作用,并且还在精神分裂症的临床前模型中具有抗精神病效应。因此,一方面,4型化合物对于治疗神经元病症特别有效,所述神经元病症诸如阿尔茨海默病、亨廷顿病、肌萎缩侧索硬化(ALS)、帕金森病、犬认知功能障碍综合征(CCDS)、雷维小体病、门克斯病、威尔逊病、克雅氏病、法尔病、与脑循环有关的急性或慢性病症诸如缺血性或出血性中风或其它的大脑出血性损伤、年龄相关的记忆损害(AAMI)、轻度认知功能损害(MCI)、与创伤有关的轻度认知功能损害(MCI)、脑震荡后综合征、创伤后应激障碍和辅助化学疗法、创伤性脑损伤(TBI)、神经元死亡介导的眼部疾病、黄斑变性、年龄有关的黄斑变性、孤独症(包括孤独症谱系障碍)、阿斯珀格综合征和雷特综合征、撕脱损伤、脊髓损伤、重症肌无力、吉兰巴雷综合征、多发性硬化、糖尿病性神经病、纤维肌痛、与脊髓损伤相关的神经病、精神分裂症、双相性精神障碍、精神病、焦虑和抑郁。在另一个方面,4型化合物对治疗神经递质-介导的障碍特别有用,其包括但不限于脊髓损伤、糖尿病性神经病、变应性疾病和牵涉于老化保护活性的疾病,诸如年龄相关的掉发(脱发)、年龄相关的体重减轻和年龄相关的视觉障碍(白内障)。方法概述本文所述的化合物可用于在个体诸如人中治疗、预防以下各项、延缓其发作和/或延缓其发展:认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症。在一方面,本文所述的化合物可用于治疗、预防认知障碍,延缓其发作和/或延缓其发展。在另一个方面,本文所述的化合物可用于治疗、预防精神病性精神障碍,延缓其发作和/或延缓其发展。在又一个方面,本文所述的化合物可用于治疗、预防神经递质-介导的障碍,延缓其发作和/或延缓其发展。在一个(实施方案)中,神经递质-介导的障碍包括脊髓损伤、糖尿病性神经病、变应性疾病(其包括食物过敏症)和牵涉于老化保护活性的疾病,诸如年龄相关的掉发(脱发)、年龄相关的体重减轻和年龄相关的视觉障碍(白内障)。在另一个实施方案中,神经递质-介导的障碍包括脊髓损伤、糖尿病性神经病、纤维肌痛和变应性疾病(其包括食物过敏症)。在又一个(实施方案)中,神经递质-介导的障碍包括阿尔茨海默病、帕金森病、孤独症、吉兰巴雷综合征、轻度认知功能损害、多发性硬化、中风和创伤性脑损伤。在又一个(实施方案)中,神经递质-介导的障碍包括精神分裂症、焦虑、双相性精神障碍、精神病和抑郁。在另一个方面,本文所述的化合物可用于治疗、预防神经元病症,延缓其发作和/或延缓其发展。在一方面,本文所述的化合物还可用于治疗、预防以下各项、延缓其发作和/或延缓其发展:认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症,其中调节胺能G蛋白偶联受体被认为对其有益或是对其有益的。本发明还提供了改善认知功能和/或降低精神病效应的方法,其包括向有需要的个体施用对改善认知功能和/或降低精神病效应有效的量的发明化合物或其可药用盐。本发明还提供了在个体中刺激神经突向外生长和/或促进神经发生和/或增强神经营养作用的方法,其包括向有需要的个体施用对刺激神经突向外生长和/或促进神经发生和/或增强神经营养作用有效的量的本发明化合物或其可药用盐。本发明还包括刺激调节胺能G蛋白偶联受体的方法,其包括向有需要的个体施用对调节胺能G蛋白偶联受体有效的量的本发明化合物或其可药用盐。应理解本文所述的方法还包括施用包含本发明化合物的组合物的方法。用于治疗、预防认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症,延缓其发作和/或延缓其发展的方法在一方面,本发明提供了用于治疗、预防认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症,延缓其发作和/或延缓其发展的方法,其中调节胺能G蛋白偶联受体被认为对其有益或是对其有益的,所述方法包括向有需要的个体施用本发明化合物。在一些实施方案中,调节肾上腺素能受体α1D、α2A、α2B、血清素受体5-HT2A、5-HT6、5HT7、组胺受体H1和/或H2被预期有益于或是有益于认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症。在一些实施方案中,调节肾上腺素能受体α1D、α2A、α2B和血清素受体5-HT6受体被预期有益于或是有益于认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症。在一些实施方案中,调节肾上腺素能受体α1D、α2A、α2B和血清素受体5-HT6受体和调节一种或多种以下的受体:血清素5-HT7、5-HT2A、5-HT2C和组胺H1和H2被预期有益于或是有益于认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症。在一些实施方案中,调节多巴胺受体D2L被预期有益于或是有益于认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症。在某些实施方案中,调节多巴胺D2L受体和血清素受体5-HT2A被预期有益于或是有益于认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症。在一些实施方案中,通过施用1型化合物治疗、预防认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症和/或延缓其发作或发展。在一些实施方案中,通过施用2型化合物治疗、预防认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症和/或延缓其发作或发展。在一些实施方案中,通过施用3型化合物治疗、预防认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症和/或延缓其发作或发展。在一些实施方案中,通过施用4型化合物治疗、预防认知障碍、精神病性精神障碍、神经递质-介导的障碍和/或神经元病症和/或延缓其发作或发展。改善认知功能和/或降低精神病性效应的方法本发明提供了通过向有需要的个体施用本发明化合物来改善认知功能的方法。在一些实施方案中,需要或预期需要调节肾上腺素能受体α1D、α2A、α2B、血清素受体5-HT2A、5-HT6、5HT7、组胺受体H1和/或H2中的一种或多种以改善认知功能。在一些实施方案中,需要或预期需要调节α1D、α2A、α2B肾上腺素能受体和血清素5-HT6受体以改善认知功能。在一些实施方案中,需要或预期需要调节α1D、α2A、α2B肾上腺素能受体和血清素受体5-HT6和调节一种或多种以下的受体:血清素受体5-HT7、5-HT2A、5-HT2C和组胺受体H1和H2,以改善认知功能。在另一个方面,本发明包括通过向有需要的个体施用本发明化合物以降低精神病性效应的方法。在一些实施方案中,需要或预期需要调节多巴胺D2L受体以降低精神病性效应。在一些实施方案中,需要或预期需要调节多巴胺D2L受体和血清素5-HT2A受体以降低精神病性效应。在一些实施方案中,向有需要的个体施用1型化合物。在一些实施方案中,向有需要的个体施用2型化合物。在一些实施方案中,向有需要的个体施用3型化合物。在一些实施方案中,向有需要的个体施用4型化合物。刺激神经突向外生长、促进神经发生和/或增强神经营养作用的方法在另一个方面,本发明提供了刺激神经突向外生长和/或增强神经发生和/或增强神经营养作用的方法,其包括在足以刺激神经突向外生长和/或增强神经发生和/或增强神经营养作用的情况下向有需要的个体施用本发明化合物或其可药用盐。在一些实施方案中,本发明化合物以约1μM的效价强度刺激神经突向外生长,如在适合的试验诸如本文所述的试验中所测定的那样。在一些实施方案中,本发明化合物以约500nM的效价强度刺激神经突向外生长,如在适合的试验诸如本文所述的试验中所测定的那样。在一些实施方案中,本发明化合物以约50nM的效价强度刺激神经突向外生长,如在适合的试验诸如本文所述的试验中所测定的那样。在一些实施方案中,本发明化合物以约5nM的效价强度刺激神经突向外生长,如在适合的试验诸如本文所述的试验中所测定的那样。在一些实施方案中,本发明化合物是1型化合物。在一些实施方案中,本发明化合物2型化合物。在一些实施方案中,本发明化合物3型化合物。在一些实施方案中,本发明化合物4型化合物。调节胺能G蛋白偶联受体的方法本发明还涉及用于调节胺能G蛋白偶联受体活性的方法,其包括在足以调节胺能G蛋白偶联受体的活性的情况下施用本发明化合物或其可药用盐。在一些实施方案中,本发明化合物是1型化合物。在一些实施方案中,本发明化合物2型化合物。在一些实施方案中,本发明化合物3型化合物。在一些实施方案中,本发明化合物4型化合物。在一些实施方案中,胺能G蛋白偶联受体是α1D、α2A、α2B肾上腺素能受体和血清素5-HT6受体。在一些实施方案中,胺能G蛋白偶联受体是α1D、α2A、α2B肾上腺素能受体和血清素5-HT6和5-HT7受体。在一些实施方案中,胺能G蛋白偶联受体是α1D、α2A、α2B肾上腺素能受体、血清素5-HT6和一种或多种以下的受体:血清素5-HT-7、5-HT2A和5-HT2C和组胺H1和H2受体。在一些实施方案中,胺能G蛋白偶联受体是多巴胺D2L受体。在一些实施方案中,胺能G蛋白偶联受体是多巴胺D2L受体和血清素5-HT2A受体。在一些实施方案中,胺能G蛋白偶联受体是组胺H1受体。另外的方法在一方面,化合物162x、177x、150x、174x、155x、184x、171x、133x、131x、132x、170x、151x、141x、169x、172x、173x或168x用于治疗、预防认知障碍,延缓其发作和/或延缓其发展。在一个实施方案中,化合物162x、177x、150x、174x、155x、184x、171x、133x、131x、132x、170x、151x、141x、169x、172x、173x或168x用于治疗、预防精神病性精神障碍,延缓其发作和/或延缓其发展。在又一个实施方案中,化合物162x、177x、150x、174x、155x、184x、171x、133x、131x、132x、170x、151x、141x、169x、172x、173x或168x用于治疗、预防神经元病症,延缓其发作和/或延缓其发展。在又一个实施方案中,化合物162x、177x、150x、174x、155x、184x、171x、133x、131x、132x、170x、151x、141x、169x、172x、173x或168x用于治疗、预防脊髓损伤、糖尿病性神经病和牵涉于老化保护活性的疾病,延缓其发作和/或延缓其发展。在另一个方面,化合物152x、137x、139x、159x或165x用于治疗、预防认知障碍,延缓其发作和/或延缓其发展。在一个实施方案中,化合物152x、137x、139x、159x和165x用于治疗、预防精神病性精神障碍,延缓其发作和/或延缓其发展。在又一个实施方案中,化合物152x、137x、139x、159x和165x用于治疗、预防神经元病症,延缓其发作和/或延缓其发展。在又一个实施方案中,化合物152x、137x、139x、159x和165x用于治疗、预防脊髓损伤、糖尿病性神经病和/或牵涉于老化保护活性的疾病,延缓其发作和/或延缓其发展。在另一个方面,化合物167用于治疗、预防认知障碍,延缓其发作和/或延缓其发展。在一个实施方案中,化合物167用于治疗、预防精神病性精神障碍,延缓其发作和/或延缓其发展。在又一个实施方案中,化合物167用于治疗、预防神经元病症,延缓其发作和/或延缓其发展。在又一个实施方案中,化合物167用于治疗、预防除经血清素介导的疾病之外的神经递质介导的疾病,延缓其发作和/或延缓其发展。在另一个方面,化合物188x、190x或199x用于治疗、预防认知障碍,延缓其发作和/或延缓其发展。在一个实施方案中,化合物188x、190x和199x用于治疗、预防精神病性精神障碍,延缓其发作和/或延缓其发展。在又一个实施方案中,化合物188x、190x和199x用于治疗、预防神经递质-介导的障碍,延缓其发作和/或延缓其发展。在另一个实施方案中,化合物188x、190x和199x用于治疗、预防神经元病症,延缓其发作和/或延缓其发展。在另一个方面,化合物134x用于治疗、预防脊髓损伤、糖尿病性神经病、与神经损伤相关的神经病、撕脱伤、重症肌无力、吉兰巴雷综合征、多发性硬化和纤维肌痛,延缓其发作和/或延缓其发展。在另一个方面,化合物178x用于治疗、预防脊髓损伤、变应性疾病和牵涉于老化保护活性的疾病,延缓其发作和/或延缓其发展。在一个实施方案中,化合物178x用于治疗、预防撕脱伤、重症肌无力、吉兰巴雷综合征、多发性硬化和纤维肌痛,延缓其发作和/或延缓其发展。在另一个方面,化合物140x用于治疗、预防脊髓损伤、牵涉于老化保护活性的疾病、撕脱伤、重症肌无力、吉兰巴雷综合征、多发性硬化和/或纤维肌痛,延缓其发作和/或延缓其发展。在另一个方面,化合物56x用于治疗、预防脊髓损伤、变应性疾病、撕脱伤、重症肌无力、吉兰巴雷综合征、多发性硬化和纤维肌痛,延缓其发作和/或延缓其发展。通用合成方法可用许多方法制备本发明化合物,这些方法在下文中进行概述并在下文的实施例中更具体地描述。除非另有指明,在以下的方法描述中,当用于所述的式中时的各符号应为理解为表示上文所述的涉及式(I)或其实施方案的那些基团。当需要得到化合物的特定的对映体时,可使用任何适合的常规的用于分离或拆分对映体的方法,由相应的对映体的混合物获得。因此,例如,可通过对映体的混合物例如外消旋物和适合的手性化合物的反应制备非对映异构衍生物。然后可通过任何方便的方法例如通过结晶分离非对映体,并回收需要的对映体。在另一个拆分方法中,外消旋物可使用手性高效液相色谱法分离。或者,如果需要,则可通过在所述方法之一中使用适合的手性中间体得到特定的对映体。当需要得到化合物的特定的异构体或纯化反应的产物时,还可以对中间体或最终产物使用色谱法、重结晶及其他常规的分离方法。本文使用以下的缩写:薄层色谱法(TLC);小时(h);乙醇(EtOH);二甲基亚砜(DMSO);N,N-二甲基甲酰胺(DMF);三氟乙酸(TFA);四氢呋喃(THF);当量浓度(N);含水(aq.);甲醇(MeOH);二氯甲烷(DCM);保留因子(Rf)。虽然本文以实施例的方式给出某些溶剂、温度及反应时间,但其它的变型是可能的且本领域的技术人员例如基于原料的选择会优化这些条件。通用方法1表示的是在本发明化合物的合成中所使用的中间体的合成方法。通用方法1.将化合物A(1当量)和化合物B(0.76-1.4当量)在适合的溶剂诸如EtOH中混合,并在80℃加热达16小时(过夜),之后在真空下除去溶剂。碱化得到的残余物,例如用饱和的NaHCO3水溶液。用二氯甲烷萃取水层,并将合并的有机层经Na2SO4干燥,在真空下浓缩,并纯化,例如,经硅胶色谱(230-400目)使用适合的溶剂梯度诸如甲醇-二氯甲烷梯度或乙酸乙酯-己烷梯度洗脱来纯化。通用方法2中显示合成某些本发明化合物的方法。通用方法2.将化合物C(1当量)、化合物D(2-20当量)和NaH(1-20当量)在DMSO中在100-120℃加热72小时或在DMF中在120-200℃加热5-24小时。将混合物冷却至25℃,通过小心加入甲醇或水淬灭,并在真空下蒸发至干燥。将得到的粗品产物,例如,通过经硅胶色谱(100-200目或230-400目)使用甲醇-二氯甲烷梯度洗脱而纯化和/或经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化。通用方法3中显示合成本发明化合物的方法。通用方法3.将化合物C(1当量)、化合物F(2-20)、钠(0.2-0.8当量)和CuSO4(催化的)在EtOH中在120℃加热16小时。将该混合物在真空下蒸发至干燥。加入水,并将该混合物用乙酸乙酯萃取,经无水硫酸钠干燥,并浓缩。将得到的粗品产物经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化和/或经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱来纯化。通用方法4中显示合成某些本发明化合物的方法。通用方法4.将化合物C(1当量)、化合物H(2-10当量)和NaH(3当量)在DMF中在120℃加热16小时。将混合物冷却至25℃,通过小心地加入甲醇或水淬灭,并在真空下蒸发至干燥。将得到的粗品产物通过经硅胶色谱(100-200目或230-400目)使用甲醇-二氯甲烷梯度洗脱来纯化和/或经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化。通用方法5中显示合成某些本发明化合物的方法。通用方法5.将化合物C(1当量)和化合物J(1.8-2.5当量)在二异丙基胺中在80℃加热12小时。用1NNaOH碱化该混合物,并用乙酸乙酯萃取。将有机层经硫酸钠干燥,并浓缩。将得到的粗品产物经硅胶色谱(230-400目)使用甲醇-二氯甲烷梯度洗脱,随后经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化。通用方法6中显示合成某些本发明化合物的方法。通用方法6.将化合物C(1当量)、化合物L(4-7.5当量)和NaH(3当量)在DMF中在120℃加热16小时。通过甲醇淬灭该混合物,并蒸发至干燥。将得到的粗品产物经硅胶色谱(230-400目)使用甲醇-二氯甲烷梯度洗脱,随后经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化。通用方法7中显示合成某些本发明化合物的方法。通用方法7.将在DMF:H2O(2:1)中的化合物C(1当量)、化合物N(2.05当量)和K3PO4(1.9当量)用氮气净化30分钟,之后加入PdCl2(PPh3)2(25mg,0.04mmol),并将该混合物在100℃加热4小时。在真空下浓缩该混合物,并将粗品产物经硅胶色谱(230-400目)纯化,得到化合物O。将化合物O(1当量)、2-甲基-5-乙烯基吡啶(12-15当量)和NaH(3-4当量)在DMSO中在100℃加热48小时。通过甲醇淬灭该混合物,并蒸发至干燥。将得到的粗品产物经硅胶色谱(230-400目)使用甲醇-二氯甲烷梯度洗脱而纯化,得到化合物Q。通用方法8中显示合成本发明化合物的方法。通用方法8.将化合物A(1当量)、三乙胺(1当量)和化合物R(1当量)溶于EtOH中,并在80℃加热2小时,之后加入化合物B(1至1.5当量)。并将该混合物在80℃再加热16小时。在真空下除去溶剂。用EtOAc稀释得到的残余物,并用饱和的NaHCO3水溶液洗涤。用乙酸乙酯萃取水层两次,并经Na2SO4干燥合并的有机层,并浓缩。将得到的粗品产物经硅胶色谱(100-200目或230-400目)使用甲醇-二氯甲烷梯度洗脱、经中性氧化铝使用乙酸乙酯-己烷梯度洗脱和/或经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化。通用方法9中显示合成本发明化合物的方法。通用方法9.概括而言,可将一个被适合地取代的肼U与被适当地取代的卤代烷V反应生成被取代的肼W,其中肼的内部氮被取代,如上文所示。中间体W与被适当地取代的环己酮X反应将生成由通式结构Y所描述的类型的结构。通用方法10中显示合成某些本发明化合物的方法。通用方法10.概括而言,可将一个被适合地取代的肼U与被适当地取代的卤代烷V反应生成被取代的肼W,其中肼的内部氮被取代,如上文所示。中间体W与被适当地取代的4-二烷基氨基环己酮Z反应,其中Ra和Rb如上文对“被取代的氨基”所定义,将生成由结构AA所概述的类型的结构。同样,使用被适当地取代的3-二烷基氨基环己酮将生成由通式结构AB和AC所描述的类型的化合物。参考:JournalofMedicinalChemistry,1977,第20卷,第4期,第487-492页。通用方法11显示的是在某些本发明化合物的合成中所使用的中间体的合成方法。通用方法11.参见:HelveticaChimicaActa,2001,第84卷,第2347-2354页。通用方法12中显示合成某些本发明化合物的方法。通用方法12.以上所示的N-(2-氧代乙基)酰胺与劳森试剂(2,4-二(4-甲氧基苯基)-1,3,2,4-二硫杂二磷杂环丁烷-2,4-二硫化物)反应将生成式Ga化合物。参见:HelveticaChimicaActa,2001,第84卷,第2347-2354页。通用方法13中显示合成某些本发明化合物的方法。通用方法13.将以上所示的酰基氯与氨基硫脲(包含R13b)反应,随后在碳酸氢钠水溶液的存在下加热,将生成式Gb化合物。通用方法14中显示合成某些本发明化合物的方法。通用方法14.将以上所示的酰肼与不同的异硫氰酸烷基酯(包含R13b)反应,随后在碳酸氢钠水溶液的存在下加热,将生成式Gb化合物。参见:JournalofMedicinalChemistry,1994,第37卷,第125-132页。通用方法15中显示合成某些本发明化合物的方法。通用方法15.将以上所示的酰肼与酰基氯(烷酰氯、芳酰氯或杂芳酰氯)反应,将生成式Gc化合物。参见:Synlett,2005,第17期,第2595-2598页。通用方法16中显示合成某些本发明化合物的方法。通用方法16.将以上所示的羟基脒与不同的芳族酸和杂芳族酸反应,将生成式Gd化合物。参见:TetrahedronLetters,2006,47,第2965-2967页。通用方法17中显示合成某些本发明化合物的方法。通用方法17.将约1:1的原料701和702的混合物在含酸的醇溶剂例如乙醇(例如5%氯化氢的乙醇溶液)中在回流下进行反应,将生成Ga化合物。Hal是卤素,例如,氯或溴,且R1、R19、R13a和R14a如上文所定义。通用方法18中显示合成某些本发明化合物的方法。通用方法18.将约1:1的原料801和802的混合物在醇溶剂例如甲醇或乙醇中在回流下进行反应,将得到中间体化合物诸如803。随后在约180-270℃加热803将生成式Gc化合物。Alk是烷基,例如,甲基或乙基,且R1、R19和R13c如上文所定义。通用方法19中显示合成某些本发明化合物的方法。通用方法19.将1:1:1的原料901、902的混合物例如R13dCOOEt和903(例如乙醇钠)在醇溶剂例如乙醇中在回流下进行反应,将得到式Gd化合物。Alk是烷基,例如,甲基或乙基,且R1、R19和R13d如上文所定义。通用方法20中显示合成某些本发明化合物的方法。通用方法20.将约1:1-1:3的原料901和1001的混合物在碱性有机溶剂例如吡啶中在回流下反应,将提供式Gd化合物。R1,R19和R13d如上文所定义。通用方法21中显示合成某些本发明化合物的方法。通用方法21.依据通用方法21合成化合物59H、61H、90H和109H。通用方法22中显示合成某些本发明化合物的方法。通用方法22.依据通用方法22合成化合物222H、223H、234H和242H。通用方法23中显示合成某些本发明化合物的方法。通用方法23.依据通用方法23化合物合成9H、38H、39H和40H。通用方法24中显示合成某些本发明化合物的方法。通用方法24.依据通用方法24合成化合物151H、218H、219H和220H。如本领域技术人员所知,可修改上文所述的方法。下文的实施例中提供了每个通用方法的特定的实施例。以下实施例用于阐明但不限制本发明。本文所公开的所有参考文献的全部内容被引入本文作为参考。实施例实施例1.2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备.依据通用方法1制备2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚。具体而言,将4-甲基苯基肼盐酸盐(5g,31.5mmol)和N-甲基-4-哌啶酮盐酸盐(3.6g,24mmol)混合在EtOH(150mL)中,并在80℃加热16小时(过夜),之后在真空下除去溶剂。将得到的残余物用饱和的NaHCO3水溶液碱化。用二氯甲烷萃取水层,并经Na2SO4干燥合并的有机层,在真空下浓缩,并经硅胶色谱(230-400目)使用甲醇-二氯甲烷梯度洗脱而纯化,得到4.1g(65%收率)的2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚。TLC(Merck硅胶60F-254)Rf=0.1(10%MeOH的DCM)。实施例2.8-溴-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备.使用在EtOH(400mL)中的4-溴苯基肼盐酸盐(10g,44.7mmol)和N-甲基-4-哌啶酮盐酸盐(9.3g,62.6mmol)依据通用方法1制备8-溴-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚,纯化后得到2.5g(21%收率)的8-溴-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚。实施例3.2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备.使用在EtOH(200mL)中的苯基肼盐酸盐(5g,34.6mmol)和N-甲基-4-哌啶酮盐酸盐(7.2g,48.4mmol)依据通用方法1制备2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚,纯化后得到3.75g(58.3%收率)的2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚。实施例4.8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备.使用在EtOH(400mL)中的4-氯苯基肼盐酸盐(10g,55.8mmol)和N-甲基-4-哌啶酮盐酸盐(11.68g,78.19mmol)依据通用方法1制备8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚,纯化后得到3.1g(25%收率)的8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚。实施例5.2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备.使用在EtOH(150mL)中的苯基肼盐酸盐(3.4g,23.5mmol)和N-乙基-4-哌啶酮盐酸盐(5.3g,32.9mmol)依据通用方法1的制备2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚,纯化后得到1.4g(30%收率)的2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚。实施例6.2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚的制备.使用在EtOH(150mL)中的4-甲基苯基肼盐酸盐(5g,31.5mmol)和N-乙基-4-哌啶酮盐酸盐(4g,24.5mmol)依据通用方法1制备2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚,纯化后得到4.8g(71%收率)的2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚。实施例7.8-溴-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯的制备.使用在EtOH(10mL)中的4-溴苯基肼盐酸盐(447mg,2mmol)和N-乙酯基-4-哌啶酮(342mg,2mmol)依据通用方法1制备8-溴-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯,纯化后得到423mg(65%收率)的8-溴-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯。实施例8.8-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯的制备.使用在EtOH(10mL)中的4-甲基苯基肼盐酸盐(317mg,2mmol)和N-乙酯基-4-哌啶酮(342mg,2mmol)依据通用方法1制备8-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯,纯化后得到340mg(65%收率)的8-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯。实施例9.2,3,4,5-四氢-2,8-二甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物25)依据通用方法2制备2,3,4,5-四氢-2,8-二甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚。将2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(参见实施例1)(200mg,1mmol)、4-甲基苯乙烯(239mg,2.3mmol)和NaH(120mg,60%在油中的分散体,3mmol)在DMSO(4ml)中在120℃加热过夜(16小时),之后加入甲醇,并将该混合物浓缩至干燥。将得到的粗品产物经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化和/或经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱纯化,得到20mg(6.2%收率)的2,3,4,5-四氢-2,8-二甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例10.8-溴-3,4-二氢-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯的制备(化合物18)依据通用方法2制备8-溴-3,4-二氢-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯。由8-溴-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯(参见实施例7)(25mg,0.056mmol)、2-甲基-5-乙烯基吡啶(0.12mL)和NaH(10mg,60%在油中的分散体,0.25mmol)在DMSO(0.3ml)中制备标题化合物,纯化后得到4.5mg8-溴-3,4-二氢-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯。实施例11.3,4-二氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯的制备(化合物24)依据通用方法2制备标题化合物。通过8-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯(参见实施例8)(257mg,1mmol)、苯乙烯(239mg,2.3mmol)和NaH(120mg,60%在油中的分散体,3mmol)在DMSO(5ml)中在120℃过夜(16小时)制备3,4-二氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯,纯化后得到15mg3,4-二氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯。实施例12.2-乙基-2,3,4,5-四氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚的制备(化合物37)依据通用方法2制备标题化合物。将2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例6)(100mg,0.46mmol)、苯乙烯(1mL,8.41mmol)和NaH(200mg,60%在油中的分散体,8.30mmol)在DMF(3ml)中在200℃加热5小时制备2-乙基-2,3,4,5-四氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚,得到15mg(4.7%收率)的2-乙基-2,3,4,5-四氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚。实施例13.2-乙基-2,3,4,5-四氢-8-甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物26)依据通用方法2制备标题化合物。将2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例6)(100mg,0.4mmol)、4-甲基苯乙烯(1mL,7.6mmol)和NaH(100mg,60%在油中的分散体,2.5mmol)在DMF(2ml)中在180℃加热24小时制备2-乙基-2,3,4,5-四氢-8-甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚,纯化后得到13mg2-乙基-2,3,4,5-四氢-8-甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚。实施例14.2,3,4,5-四氢-2-甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物1)依据通用方法2制备标题化合物。由2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例3)(200mg,1.07mmol)、4-甲基苯乙烯(1.41mL,10.7mmol)和NaH(250mg,60%在油中的分散体,6.25mmol)在DMF(6ml)中在200℃加热16小时制备2,3,4,5-四氢-2-甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚,纯化后得到7mg2,3,4,5-四氢-2-甲基-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚。实施例15.2,3,4,5-四氢-2-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚的制备(化合物2)依据通用方法2制备标题化合物。由2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(200mg,1.07)、苯乙烯(1.23mL,10.65mmol)和NaH(250mg,6.25mmol)在DMF(6ml)中在200℃加热16小时制备2,3,4,5-四氢-2-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚,纯化后得到15mg2,3,4,5-四氢-2-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚。实施例16.2-乙基-2,3,4,5-四氢-5-苯乙基-1H-吡啶并[4,3-b]吲哚的制备(化合物4)依据通用方法2制备标题化合物。由2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(参见实施例5)(100mg,0.5mmol)、苯乙烯(1mL,8.64mmol)和NaH(200mg,5mmol)在DMF(4ml)中在150℃加热16小时制备2-乙基-2,3,4,5-四氢-5-苯乙基-1H-吡啶并[4,3-b]吲哚,纯化后得到15mg2-乙基-2,3,4,5-四氢-5-苯乙基-1H-吡啶并[4,3-b]吲哚。实施例17.2-乙基-2,3,4,5-四氢-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物3)依据通用方法2制备标题化合物。由2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(参见实施例5)(100mg,0.5mmol)、4-甲基苯乙烯(1mL,6.72mmol)和NaH(200mg,5mmol)在DMF(4ml)中在150℃加热16小时制备2-乙基-2,3,4,5-四氢-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚,纯化后得到16mg2-乙基-2,3,4,5-四氢-5-(4-甲基苯乙基)-1H-吡啶并[4,3-b]吲哚。实施例18.2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物31)依据通用方法2制备标题化合物。由2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例6)(214mg,1mmol)、2-甲基-5-乙烯基吡啶(1mL,2.3mmol)和NaH(120mg,3mmol)在DMSO(4ml)中在120℃加热48小时制备2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚,纯化后得到10mg2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚。实施例19.5-(4-氯苯乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物32)依据通用方法2制备标题化合物。由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(参见实施例1)(500mg,2.5mmol)、4-氯苯乙烯(3.18mL,25mmol)和NaH(300mg,7.5mmol)在DMF(10ml)中在180℃加热过夜(16小时)制备5-(4-氯苯乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚,纯化后得到15mg5-(4-氯苯乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚。实施例20.2,3,4,5-四氢-2,8-二甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物21)依据通用方法3制备标题化合物。将2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(参见实施例1)(400mg,2mmol)、2-乙烯基吡啶(500mg,5mmol)、钠(30mg,1.3mmol)和CuSO4(20mg,催化的)在EtOH(8mL)中在150℃加热过夜(16小时)。将该混合物在真空下蒸发至干燥。加入水,并用乙酸乙酯萃取该混合物,经无水硫酸钠干燥,并浓缩。将得到的粗品产物经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到15mg2,3,4,5-四氢-2,8-二甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例21.2,3,4,5-四氢-2,8-二甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物22)依据通用方法3制备标题化合物。由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(参见实施例1)(100mg,0.5mmol)、4-乙烯基吡啶(120mg,1.14mmol)和钠(5mg,0.21mmol)在乙醇(1mL)中在120℃加热16小时制备2,3,4,5-四氢-2,8-二甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚,经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到4.5mg2,3,4,5-四氢-2,8-二甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚。实施例22.2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物23)依据通用方法3制备标题化合物。由2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例6)(214mg,1mmol)、2-乙烯基吡啶(0.25ml,2.3mmol)、钠(15mg,0.65mmol)和CuSO4(10mg,催化的)在乙醇(2mL)中在120℃加热48小时制备2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到15mg2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例23.2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物27)依据通用方法3制备标题化合物。由2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例6)(214mg,1mmol)、4-乙烯基吡啶(0.25ml,2.3mmol)、钠(15mg,0.65mmol)和CuSO4(10mg,催化的)在乙醇(2mL)中在120℃加热过夜(16小时)制备2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到6mg2-乙基-2,3,4,5-四氢-8-甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例24.2-乙基-2,3,4,5-四氢-5-(4-吡啶基)-1H-吡啶并[4,3-b]吲哚的制备(化合物5)依据通用方法3制备标题化合物。由2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(参见实施例5)(200mg,1mmol)、4-乙烯基吡啶(0.26mg,2.5mmol)和钠(15mg,0.65mmol)在乙醇(2mL)中在150℃加热过夜(16小时)制备2-乙基-2,3,4,5-四氢-5-(4-吡啶基)-1H-吡啶并[4,3-b]吲哚,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到10mg2-乙基-2,3,4,5-四氢-5-(4-吡啶基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例25.2-乙基-2,3,4,5-四氢-5-(2-吡啶基)-1H-吡啶并[4,3-b]吲哚的制备(化合物6)依据通用方法3制备标题化合物。由2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(参见实施例5)(200mg,1mmol)、2-乙烯基吡啶(0.26mg,2.5mmol)、钠(15mg,0.65mmol)和CuSO4(10mg,催化的)在乙醇(2mL)中在150℃加热16小时制备2-乙基-2,3,4,5-四氢-5-(2-吡啶基)-1H-吡啶并[4,3-b]吲哚,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到8mg2-乙基-2,3,4,5-四氢-5-(2-吡啶基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例26.2,3,4,5-四氢-2-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物26)依据通用方法3制备标题化合物。由2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例3)(200mg,1mmol)、2-乙烯基吡啶(0.26mg,2.5mmol)、钠(5mg,0.21mmol)和CuSO4(5mg,催化的)在乙醇(4mL)中在120℃加热16小时(过夜)制备2,3,4,5-四氢-2-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到60mg2,3,4,5-四氢-2-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例27.8-氯-2,3,4,5-四氢-2-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物30)依据通用方法3制备标题化合物。由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例4)(220mg,1mmol)、2-乙烯基吡啶(1ml,18mmol)、钠(20mg,0.87mmol)和CuSO4(20mg,催化的)在乙醇(5mL)中在150℃加热16小时(过夜)制备8-氯-2,3,4,5-四氢-2-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,随后经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到38mg8-氯-2,3,4,5-四氢-2-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚。依据实施例27中所述的方法制备化合物54,其中使用被适当地取代的试剂:在乙醇中的8-氯-2,3,4,5-四氢-2-乙基-1H-吡啶并[4,3-b]吲哚、2-乙烯基吡啶、钠和CuSO4。实施例28.2,3,4,5-四氢-2-甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物10)依据通用方法3制备标题化合物。由2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例3)(200mg,1.07mmol)、4-乙烯基吡啶(0.26mL,2.41mmol)、钠(5mg,0.21mmol)和CuSO4(5mg,催化的)在乙醇(4mL)在120℃加热16小时(过夜)制备2,3,4,5-四氢-2-甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到60mg2,3,4,5-四氢-2-甲基-5-(2-(吡啶-4-基)乙基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例29.2,3,4,5-四氢-2,8-二甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚的制备(化合物139)依据通用方法4制备标题化合物。由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(参见实施例1)(200mg,1mmol)、5-(氯甲基)-2-甲基吡啶(324mg,2.3mmol)和NaH(120mg,3mmol)在DMF(4ml)中在120℃加热16小时制备2,3,4,5-四氢-2,8-二甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚。通过小心地加入甲醇或水将该反应淬灭,并将该混合物在真空下蒸发至干燥。将得到的粗品产物经硅胶色谱(100-200目或230-400目)通过甲醇-二氯甲烷梯度洗脱而纯化并经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到12.4mg2,3,4,5-四氢-2,8-二甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例30.2-乙基-2,3,4,5-四氢-8-甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚的制备(化合物138)依据通用方法4制备标题化合物。将2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例6)(214mg,1mmol)、5-(氯甲基)-2-甲基吡啶(324mg,2.3mmol)和NaH(120mg,3mmol)在DMF(4ml)中在120℃加热16小时,经硅胶色谱(100-200目或230-400目)使用甲醇-二氯甲烷梯度洗脱而纯化后,得到50mg2-乙基-2,3,4,5-四氢-8-甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚。实施例31.2,3,4,5-四氢-2-甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚的制备(化合物17)依据通用方法4制备标题化合物。将2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例3)(186mg,1mmol)、5-(氯甲基)-2-甲基吡啶(324mg,2.3mmol)和NaH(120mg,3mmol)在DMF(3ml)中在120℃加热16小时,经硅胶色谱(230-400目)使用甲醇-二氯甲烷梯度洗脱而纯化后,得到30mg2,3,4,5-四氢-2-甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚。实施例32.5-苄基-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物137)依据通用方法5制备标题化合物。将2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(实施例1)(100mg,0.5mmol)和苄基氯(0.11ml,0.9mmol)在二异丙基胺(2mL)中在80℃加热12小时。将该混合物用1NNaOH碱化,并用乙酸乙酯萃取。有机层经硫酸钠干燥,并浓缩。将得到的粗品产物经硅胶色谱(230-400目)使用甲醇-二氯甲烷梯度洗脱而纯化,随后经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到20mg5-苄基-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例33.5-苄基-2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物140)依据通用方法5制备标题化合物。将2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例6)(100mg,0.46mmol)和苄基氯(0.11ml,0.9mmol)在二异丙基胺(2mL)中在80℃加热12小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到40mg5-苄基-2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例34.5-苄基-2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物16)依据通用方法5制备标题化合物。将2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(参见实施例5)(100mg,0.5mmol)、苄基氯(158mg,1.25mmol)在二异丙基胺(2mL)在80℃加热12小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到50mg5-苄基-2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例35.外消旋-2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-对-甲苯基乙醇的制备(化合物28)依据通用方法6制备标题化合物。将2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(参见实施例1)(2.2g,11mmol)、4-甲基苯乙烯氧化物(5.8g,44mmol)和NaH(1.3g,32.5mmol)在DMF(70mL)中在120℃加热16小时(过夜)。将该混合物通过甲醇淬灭,并蒸发至干燥。将得到的粗品产物经硅胶色谱(230-400目)使用乙酸乙酯-己烷梯度洗脱而纯化,得到1.3g外消旋-2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-对-甲苯基乙醇。通过乙醇HCl处理将该游离碱转化为其盐酸盐。实施例36.外消旋-2-(2-乙基-1,2,3,4-四氢-8-甲基吡啶并[4,3-b]吲哚-5-基)-1-对-甲苯基乙醇的制备(化合物29)依据通用方法6制备标题化合物。将2-乙基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例6)(214mg,1mmol)、4-甲基苯乙烯氧化物(1mL,7.5mmol)和NaH(120mg,3mmol)在DMF(4ml)中在120℃加热16小时(过夜),经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到50mg外消旋-2-(2-乙基-1,2,3,4-四氢-8-甲基吡啶并[4,3-b]吲哚-5-基)-1-对-甲苯基乙醇,为三氟乙酸盐。实施例37.外消旋-2-(1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-对-甲苯基乙醇的制备(化合物37)依据通用方法6制备标题化合物。将2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(参见实施例3)(400mg,2.1mmol)、4-甲基苯乙烯氧化物(2.1g,15.7mmol)和NaH(252mg,6.3mmol)在DMF(5ml)中在120℃加热16小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到75mg外消旋-2-(1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-对-甲苯基乙醇,为三氟乙酸盐。实施例38.外消旋-2-(2-乙基-1,2,3,4-四氢吡啶并[4,3-b]吲哚-5-基)-1-对-甲苯基乙醇的制备(化合物8)依据通用方法6制备标题化合物。将2-乙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(参见实施例5)(400mg,2.0mmol)、4-甲基苯乙烯氧化物(2.01g,15mmol)和NaH(240mg,6mmol)在DMF(6ml)中在120℃加热16小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到120mg外消旋-2-(2-乙基-1,2,3,4-四氢吡啶并[4,3-b]吲哚-5-基)-1-对-甲苯基乙醇,为三氟乙酸盐。实施例39.3,4-二氢-5-(2-(6-甲基吡啶-3-基)乙基)-8-(吡啶-3-基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯的制备(化合物19)依据通用方法7制备标题化合物。将8-溴-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯(参见实施例7)(100mg,0.3mmol)、吡啶-3-硼酸(76mg,0.62mmol)和K3PO4(120mg,0.57mmol)在DMF:H2O(2:1,3ml)中的混合液用氮气净化30分钟。加入PdCl2(PPh3)2(25mg,0.04mmol),并将该反应物在100℃加热4小时。将该混合物在真空下浓缩,并将粗品产物经硅胶色谱(230-400目)使用乙酸乙酯-己烷梯度洗脱而纯化,得到62mg3,4-二氢-8-(吡啶-3-基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯。将该中间体(25mg,0.078mmol)、2-甲基-5-乙烯基吡啶(0.120ml,1mmol)和NaH(10mg,60%在油中的分散体,0.02mmol)在DMSO(0.3ml)中在100℃加热48小时,之后加入甲醇,并将该混合物浓缩至干燥。将得到的粗品产物经硅胶色谱(100-200目或230-400目)使用甲醇-二氯甲烷梯度洗脱纯化,得到7mg3,4-二氢-5-(2-(6-甲基吡啶-3-基)乙基)-8-(吡啶-3-基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸乙酯。实施例40.2,3,4,5-四氢-2,8-二甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚的制备(化合物20)依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(2g,12.6mmol)、2-苯基乙基溴(1.7ml,12.6mmol)和三乙胺(1.7mL,12.6mmol)在乙醇(6ml)中在25℃搅拌1小时,之后将该混合物在80℃加热2小时。将该混合物冷却至25℃,加入N-甲基-4-哌啶酮盐酸盐(2.87g,18.9mmol),并持续在80℃加热16小时。将该混合物在真空下浓缩,通过加入饱和的NaHCO3水溶液碱化,并用乙酸乙酯萃取。经无水硫酸钠干燥有机层,并浓缩。将粗品产物经硅胶色谱(230-400目)使用乙酸乙酯-己烷梯度洗脱而纯化,得到150mg2,3,4,5-四氢-2,8-二甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚。通过用在无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例41.5-(2-氟苯乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物34)依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(500mg,3.1mmol)、2-氟苯基乙基溴(639mg,3.1mmol)、三乙胺(313mg,3.1mmol)和N-甲基-4-哌啶酮盐酸盐(692mg,4.6mmol)在乙醇(2ml)中合并,经中性氧化铝使用乙酸乙酯-己烷梯度洗脱而纯化后,得到30mg5-(2-氟苯乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚。通过用在无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例42.5-(2-环己基乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物163)依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(500mg,3.1mmol)、1-溴-2-环己基乙烷(602mg,3mmol)、三乙胺(303mg,3mmol)和N-甲基-4-哌啶酮盐酸盐(589mg,4.5mmol)在乙醇(3ml)中合并,经中性氧化铝使用乙酸乙酯-己烷梯度洗脱而纯化后,得到150mg5-(2-环己基乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚。实施例43.5-(4-氟苯乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物33)依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(312mg,1.9mmol)、4-氟苯基乙基溴(400mg,1.9mmol)、三乙胺(0.264ml,1.9mmol)和N-甲基-4-哌啶酮盐酸盐(424mg,2.8mmol)在乙醇(3ml)中合并,经硅胶色谱(100-200目或230-400目)使用乙酸乙酯-己烷梯度洗脱而纯化后,得到10mg5-(4-氟苯乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚。通过用在无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例44.2-环丙基-2,3,4,5-四氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚的制备(化合物35)依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(500mg,3.1mmol)、2-苯基乙基溴(0.43ml,3.1mmol)、三乙胺(0.43ml,3.1mmol)和N-环丙基-4-哌啶酮盐酸盐(650mg,4.72mmol)在乙醇(3ml)中合并,经中性氧化铝使用乙酸乙酯-己烷梯度洗脱而纯化后,得到62mg2-环丙基-2,3,4,5-四氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚。通过用在无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。依据通用方法8并如在实施例44中所述合成化合物51,其中使用被适当地取代的试剂:将对-甲苯基肼盐酸盐、5-(2-溴乙基)-2-甲基吡啶、三乙胺和N-环丙基-4-哌啶酮盐酸盐在乙醇中合并。实施例45.8-氯-2,3,4,5-四氢-2-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚的制备(化合物36)依据通用方法8制备标题化合物。将4-氯苯基肼盐酸盐(500mg,2.79mmol)、2-苯基乙基溴(0.38ml,2.79mmol)、三乙胺(0.39ml,2.79mmol)和N-甲基-4-哌啶酮盐酸盐(623mg,4.18mmol)在乙醇(2ml)中合并,经中性氧化铝使用乙酸乙酯-己烷梯度洗脱而纯化,随后经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到15mg8-氯-2,3,4,5-四氢-2-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。使用被适当地取代的试剂依据通用方法8并如在实施例45中所述合成化合物43、化合物81、化合物82和化合物83。化合物43试剂:将4-氯苯基肼盐酸盐、1-(2-溴乙基)-4-甲基苯、三乙胺和N-甲基-4-哌啶酮盐酸盐在乙醇中合并。化合物81试剂:将3-氯苯基肼盐酸盐、1-(2-溴乙基)-4-甲基苯、三乙胺和N-甲基-4-哌啶酮盐酸盐在乙醇中合并。化合物82试剂:将3-氯苯基肼盐酸盐、1-(2-溴乙基)-4-甲基苯、三乙胺和N-甲基-4-哌啶酮盐酸盐在乙醇中合并。化合物83试剂:将2-氯苯基肼盐酸盐、1-(2-溴乙基)-4-甲基苯、三乙胺和N-甲基-4-哌啶酮盐酸盐在乙醇中合并。实施例46.8-氯-2,3,4,5-四氢-2-甲基-5-(2-吗啉代乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物186)依据通用方法8制备标题化合物。将4-氯苯基肼盐酸盐(2g,11mmol)、4-(2-溴乙基)吗啉(2.1g,11mmol)、三乙胺(4.6ml,33mmol)和N-甲基-4-哌啶酮盐酸盐(1.6g,11mmol)在乙醇(20ml)中合并,经硅胶(230-400目)色谱用丙酮-己烷梯度洗脱而纯化后,得到180mg8-氯-2,3,4,5-四氢-2-甲基-5-(2-吗啉代乙基)-1H-吡啶并[4,3-b]吲哚。通过用在无水THF中的草酸(2当量)处理将该游离碱转化为其二草酸盐。实施例47.8-氯-5-(2-环己基乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物164)依据通用方法8制备标题化合物。将4-氯苯基肼盐酸盐(1g,5.5mmol)、1-溴-2-环己基乙烷(0.87ml,5.5mmol)、三乙胺(2.31ml,16.6mmol)和N-甲基-4-哌啶酮盐酸盐(810mg,5.5mmol)在乙醇(10ml)中合并,经硅胶(230-400目)色谱用丙酮-己烷梯度洗脱而纯化后,得到16mg8-氯-5-(2-环己基乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例48.9-氯-5-(2-环己基乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(化合物187)和7-氯-5-(2-环己基乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物188)依据通用方法8制备标题化合物。将3-氯苯基肼盐酸盐(5g,27.9mmol)、1-溴-2-环己基乙烷(4.37ml,27.9mmol)、三乙胺(11.66ml,83.7mmol)和N-甲基-4-哌啶酮盐酸盐(4g,27.9mmol)在乙醇(30ml)中合并,经硅胶(230-400目)色谱用丙酮-己烷梯度洗脱而纯化后,得到9-氯-5-(2-环己基乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚和7-氯-5-(2-环己基乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例49.2,3,4,5-四氢-2,8-二甲基-5-(2-(吡咯烷-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物221)依据通用方法8制备标题化合物。将4-甲基苯基肼盐酸盐(2g,12.6mmol)、1-(2-溴乙基)吡咯烷氢溴酸盐(8.1g,31.5mmol)、三乙胺(17.5ml,126mmol)和N-甲基-4-哌啶酮盐酸盐(1.87g,12.6mmol)在乙醇(15ml)中合并,经硅胶(230-400目)色谱用丙酮-己烷梯度洗脱而纯化后,得到180mg2,3,4,5-四氢-2,8-二甲基-5-(2-(吡咯烷-1-基)乙基)-1H-吡啶并[4,3-b]吲哚。通过用无水THF中的草酸(2当量)处理将该游离碱转化为其二草酸盐。实施例50.3,4-二氢-3-羟基-2,8-二甲基-5-苯乙基-2H-吡啶并[4,3-b]吲哚-1(5H)-酮的制备(化合物215)依据上文概述和下文详述的合成方案制备标题化合物。实施例50A:1-苯乙基-1-对-甲苯基肼的制备.将对-甲苯基肼盐酸盐(10.0g,63mmol)混悬于EtOH(45mL)中。历经5-10分钟的时间向该混悬液中滴加三乙胺(9.0mL,63mmol)。将该反应混合液再搅拌10分钟。历经10至15分钟的时间在25℃滴加苯乙基溴(9mL,63mmol)。将该反应混合液在80℃加热2小时,这时通过TLC和LC-MS发现该反应已经完成。将该反应混合液减压浓缩,将残余物混悬于饱和的NaHCO3水溶液(pH9)中,并用乙酸乙酯(100mLx2)萃取。分离有机层,干燥(Na2SO4)并浓缩,得到粗品产物,为深褐色油状物(14g)。经快速柱色谱(硅胶,230-400目,洗脱:8-10%乙酸乙酯-己烷)纯化粗品产物,得到3.7g纯品产物,为黄色油状物。收率:26%(未优化)。实施例50B:2,5-二甲基-1-苯乙基-1H-吲哚-3-甲酸乙酯的制备.将1-苯乙基-1-对-甲苯基肼(3.7g,16.3mmol)溶于乙醇HCl(40mL,该溶液的pH是酸性的)中,并向其中加入乙酰乙酸乙酯(2.0mL,16.3mmol)。将该反应混合液在110℃加热1.5小时,这时通过TLC和LC-MS发现该反应已经完成。将该反应混合液减压浓缩,将残余物混悬于饱和的NaHCO3水溶液(pH9)中,并用乙酸乙酯(100mLx2)萃取。分离有机层,干燥(Na2SO4)并浓缩,得到粗品产物,为深褐色油状物。经快速柱色谱(硅胶,230-400目,洗脱:5-10%乙酸乙酯-己烷)纯化粗品产物,得到3.2g纯品产物,为黄色固体。实施例50C:2,5-二甲基-1-苯乙基-1H-吲哚-3-甲酸的制备.将2,5-二甲基-1-苯乙基-1H-吲哚-3-甲酸乙酯(500mg)和NaOH(500mg)在乙醇(15mL)中的混合液在100℃加热3小时,这时通过TLC和LC-MS发现该反应已经完成。通过加入乙醇HCl将该反应混合液酸化至pH4-5,并减压蒸发。将还含有在NaOH中和过程中产生的NaCl的残余物(1g)未经任何纯化用于下一步反应。实施例50D:2,5-二甲基-1-苯乙基-1H-吲哚-3-甲酰氯的制备.在0℃向所述甲酸(历经上述两个实施例的1.6g粗品产物,认为其约为2.5mmol)在无水CH2Cl2(15ml)中的搅拌的溶液中加入一滴DMF,随后通过注射器加入草酰氯(0.6mL,6.9mmol)。在0℃搅拌15分钟后,除去冰浴,并将该混合液在环境温度搅拌55分钟。将该混合液减压浓缩至干燥(避免吸湿),得到粗制的酰基氯,将其存储在氮气中,并且未经纯化地用于下一个实施例中。实施例50E:N,2,5-三甲基-1-苯乙基-1H-吲哚-3-甲酰胺的制备.将实施例50D中的酰基氯溶于CH2CI2(10ML)中,并冷却至0℃。向其中加入过量的甲胺在DCM(50ml)中的溶液。将该反应缓慢温至25℃,并在此温度另外搅拌1小时,这时通过TLC和LC-MS发现该反应已经完成。将该溶液在CH2Cl2和水之间分配。分离有机层,经Na2SO4干燥,并减压浓缩,得到650mg浅褐色固体。实施例50F:3,4-二氢-3-羟基-2,8-二甲基-5-苯乙基-2H-吡啶并[4,3-b]吲哚-1(5H)-酮的制备.将酰胺(125mg,0.41mmol)在3mL四氢呋喃中的溶液冷却至-25至-30℃(在N2下),并以使得其内部温度保持在-25至-30℃之间的速度加入n-BuLi在己烷中的溶液(1mL,1.6M,1,6mmol)。将得到的深红色溶液在-25至-30℃搅拌40分钟,并在-25至-30℃加入DMF(0.1mL)。添加完成后,将该溶液在-25至-30℃搅拌30分钟。缓慢加入盐酸(0.3mL,6N),在此期间保持温度低于5℃(pH3-4)。在真空下浓缩该混合液,得到90mg黄色半固体。用乙醚-己烷研磨该产物,随后用乙醚研磨,并过滤,得到30mg黄色固体。依据通用方法8,根据对2,3,4,5-四氢-2,8-二甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚(实施例47)的制备所述的类似的方法使用对-甲苯基肼盐酸盐、5-(2-溴乙基)-2-甲基吡啶和1-甲基哌啶-2,4-二酮制备化合物216。实施例51.(化合物47)和(化合物53)的制备依据通用方法8使用被适当地取代的试剂制备标题化合物:(化合物47)试剂:在乙醇中的4-(三氟甲基)苯基肼盐酸盐、1-(2-溴乙基)-4-甲基苯、三乙胺和N-甲基-4-哌啶酮盐酸盐,和(化合物53)试剂:在乙醇中的4-(三氟甲基)苯基肼盐酸盐、5-(2-溴乙基)-2-甲基吡啶、三乙胺和N-甲基-4-哌啶酮盐酸盐。实施例52.2,3,4,5-四氢-2,8-二甲基-5-(2-(哌啶-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物224)依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(2.0g,12.6mmol)、1-(2-氯乙基)-哌啶一盐酸盐(2.32g,12.6mmol)、三乙胺(5.3ml,37.8mmol)和N-甲基-4-哌啶酮盐酸盐(1.87g,2.1mmol)溶于乙醇(30ml)中,经硅胶(230-400目)色谱用丙酮-己烷梯度洗脱而纯化后,得到180mg2,3,4,5-四氢-2,8-二甲基-5-(2-(哌啶-1-基)乙基)-1H-吡啶并[4,3-b]吲哚。通过用无水THF中的草酸(2当量)处理将该游离碱转化为其二草酸盐。实施例53.8-氯-2,3,4,5-四氢-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物225)依据通用方法8制备标题化合物。将4-氯苯基肼盐酸盐(9g,50mmol)、5-(2-溴乙基)-2-甲基吡啶(10g,50mmol)、三乙胺(21ml,150mmol)和N-甲基-4-哌啶酮盐酸盐(7.5g,50mmol)溶于乙醇(100ml)中,经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化,随后经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到390mg8-氯-2,3,4,5-四氢-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚。实施例54.1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-4-基氨基甲酸叔丁基酯的制备(化合物226)实施例54A.3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5基)丙酸的制备.使用在乙醇(200ml)中的甲苯基肼盐酸盐(20g,126mmol)、丙酸3-溴乙基酯(22.8g,126mmol)、三乙胺(38.1g,378mmol)和N-甲基-4-哌啶酮盐酸盐(18.77g,126mmol)完成3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸乙酯的制备,经中性氧化铝色谱用二氯甲烷-己烷梯度洗脱而纯化后,得到1.5g3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸乙酯。实施例54B.3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸的制备.将3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸乙酯(参见实施例54A)(1.5g)和NaOH(3N,30ml)在乙醇(30ml)中的混合液在50℃搅拌3小时,之后将其冷却至室温,并用浓HCl中和。减压除去溶剂,得到粗制的3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸。实施例54C.如下所述得到标题化合物1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-4-基氨基甲酸叔丁基酯。将3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸(500mg,1.8mmol)(参见实施例54B)与哌啶-4-基氨基甲酸叔丁基酯(0.366ml,1.8mmol)、EDCl-HCl(0.35g,1.8mmol)和三乙胺(0.253ml,1.8mmol)在二氯甲烷(20ml)中搅拌,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱而纯化,随后经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到50mg1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-4-基氨基甲酸叔丁基酯,为三氟乙酸盐。实施例55.3,4-二氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸2,2,2-三氯乙基酯的制备(化合物227)依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(10g,63mmol)、2-苯基乙基溴(11.6g,63mmol)、三乙胺(19.4g,189mmol)和4-氧代哌啶-1-甲酸2,2,2-三氯乙基酯(452mg,2mmol)溶于乙醇(10ml)中在90℃加热3小时,经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到200mg3,4-二氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸2,2,2-三氯乙基酯。化合物235可依据实施例55中所述的方法使用被适当地取代地试剂制备。将1-(2-(6-甲基吡啶-3-基)乙基)-1-对-甲苯基肼(500mg)和N-三氯乙氧羰基-哌啶酮(560mg)在15mL乙醇HCl中混合,并搅拌20分钟。减压除去挥发物,将残余物溶于乙醇(5mL)中,并将该溶液在90℃加热3小时(通过LCMS和TLC监测该反应)。将该反应混合液冷却至25℃,并减压蒸发。用饱和的NaHCO3水溶液碱化残余物,并用乙酸乙酯萃取。经Na2SO4干燥合并的乙酸乙酯层,并减压浓缩,得到粗品产物。将粗品产物经硅胶快速色谱(洗脱:己烷至10%丙酮己烷梯度洗脱)纯化,得到100mg产物,为淡黄色油状物。TLCRf0.2,使用20%丙酮-己烷。实施例56.2,3,4,5-四氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚的制备(化合物228)将3,4-二氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸2,2,2-三氯乙基酯(100mg,0.2mmol)和Zn粉(120mg,1.9mmol)在乙酸(1.2ml)中的混合液在25℃搅拌2小时。将该反应混合液用饱和的NaHCO3水溶液碱化,并用乙酸乙酯萃取。有机层经无水硫酸钠干燥,并蒸发,经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到30mg2,3,4,5-四氢-8-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚。实施例57.2,3,4,5-四氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚N'-氧化物的制备(化合物229)在865-70℃将2,3,4,5-四氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚(98mg,3.1mmol)与过氧化氢(30%在水中,0.2ml)在冰醋酸(1.2ml)中搅拌20小时,得到76mg相应的N,N'-二氧化物。向在乙酸(0.3ml)和甲醇(2ml)中的该中间体(76mg,0.21mmol)中加入亚硫酸氢钠(40%在水中,0.2ml),并将该反应混合液在0℃搅拌0.5小时。将该反应混合液用饱和的NaHCO3水溶液碱化,并用乙酸乙酯萃取,得到50mg2,3,4,5-四氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚N'–氧化物。实施例58A.3,4-二氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸2,2,2-三氯乙基酯的制备.依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(5g,31.5mmol)、5-(2-溴乙基)-2-甲基吡啶(6.3g,31.5mmol)、三乙胺(13ml,94.5mmol)和4-氧代哌啶-1-甲酸2,2,2-三氯乙基酯(563mg,2mmol)溶于乙醇(15ml)中,经硅胶(230-400目)色谱用丙酮-己烷梯度洗脱而纯化后,得到100mg3,4-二氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸2,2,2-三氯乙基酯。实施例58B.2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备.将3,4-二氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸2,2,2-三氯乙基酯(100mg,0.2mmol)和Zn粉(120mg,1.9mmol)在乙酸(1.2ml)中的混合液在25℃搅拌12小时。将该反应混合液用饱和的氨水碱化,并用乙酸乙酯萃取。有机层经无水硫酸钠干燥,并蒸发,经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到75mg2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例59.1-(4-氨基哌啶-1-基)-3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙-1-酮的制备(化合物231)在25℃将1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-4-基氨基甲酸叔丁基酯(参见实施例54)(50mg)与三氟乙酸(2ml)在二氯甲烷(2ml)中搅拌16小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到10mg1-(4-氨基哌啶-1-基)-3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙-1-酮,为三氟乙酸盐。实施例60.4-溴-1,2-二氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-4H-吡啶并[4,3-b]吲哚-3(5H)-酮的制备(化合物232)依据通用方法8制备标题化合物。将对-甲苯基肼盐酸盐(1.27g,8mmol)、5-(2-溴乙基)-2-甲基吡啶(1.6g,8mmol)、三乙胺(3.5ml,24mmol)和1-甲基哌啶-2,4-二酮(4.2g,8mmol)溶于在乙醇(25ml)中,经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到40mg1,2-二氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-4H-吡啶并[4,3-b]吲哚-3(5H)-酮。向该中间体(25mg,0.07mmol)中加入N-溴琥珀酰胺(26mg,0.1)和在四氯化碳(10ml)中的偶氮二异丁腈(1mg),并将该反应混合液在80℃加热12小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到5mg4-溴-1,2-二氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-4H-吡啶并[4,3-b]吲哚-3(5H)-酮,为三氟乙酸盐。实施例61.2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物233)实施例61A.2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙-2-炔基)-1H-吡啶并[4,3-b]吲哚的制备.将2,3,4,5-四氢-2,8-二甲基-5-(丙-2-炔基)-1H-吡啶并[4,3-b]吲哚(通过通用方法8得到;将对-甲苯基肼盐酸盐(600mg,3.7mmol)、炔丙基溴(以重量计80%的在甲苯中的溶液,0.34ml,3.7mmol)、三乙胺(1.5ml,11.3mmol)和N-甲基-4-哌啶酮盐酸盐(316mg,2.1mmol)溶于乙醇(15ml)中,经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到80mg2,3,4,5-四氢-2,8-二甲基-5-(丙-2-炔基)-1H-吡啶并[4,3-b]吲哚)(150mg,0.6mmol)、2-溴吡啶(0.06ml,0.6mmol)、双(三苯基膦)二氯化钯(8mg,0.012mmol)、CuI(1mg,0.006mmol)和三乙胺(0.01ml,0.071mmol)在乙腈(5ml)中的混合液在80℃加热1.5小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到108mg2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙-2-炔基)-1H-吡啶并[4,3-b]吲哚。实施例61B.将2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙-2-炔基)-1H-吡啶并[4,3-b]吲哚(参见实施例61A)(20mg,0.06mmol)用10%Pd-C(10mg)在1个大气压的氢气下在甲醇(2ml)中氢化,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到5mg2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙基)-1H-吡啶并[4,3-b]吲哚。通过用乙醇HCl处理将该游离碱转化为其二HCl盐。实施例62.5-(2-氟苯乙基)-2,3,4,5-四氢-8-碘-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物89)依据通用方法8制备标题化合物。将4-碘苯基肼盐酸盐(500mg,2.1mmol)、2-氟苯乙基溴(0.3ml,2.1mmol)、三乙胺(0.8ml,6.3mmol)和N-甲基-4-哌啶酮盐酸盐(312mg,2.1mmol)溶于乙醇(10ml)中,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到5mg5-(2-氟苯乙基)-2,3,4,5-四氢-8-碘-2-甲基-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例63.1,2-二氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-4H-吡啶并[4,3-b]吲哚-3(5H)-酮的制备(化合物195)依据通用方法8,根据对2,3,4,5-四氢-2,8-二甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚(实施例47)的制备所述的相类似的方法,使用对-甲苯基肼盐酸盐、5-(2-溴乙基)-2-甲基吡啶和1-甲基哌啶-2,4-二酮制备标题化合物。实施例64.8-(三氟甲基)-2,3,4,5-四氢-2-甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚的制备(化合物80)依据通用方法8,根据对2,3,4,5-四氢-2,8-二甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚(参见实施例47)的制备所述的相类似的方法,使用4-(三氟甲基)苯基肼、2-苯基乙基溴和N-甲基-4-哌啶酮盐酸盐制备标题化合物。实施例65.5-(4-氯苯乙基)-8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物45)依据通用方法8,根据对2,3,4,5-四氢-2,8-二甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚(参见实施例47)的制备所述的相类似的方法,使用4-氯苯基肼盐酸盐、4-氯苯乙基溴和N-甲基-4-哌啶酮盐酸盐制备标题化合物。实施例66.5-(4-氟苯乙基)-8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物162)依据通用方法8,根据对2,3,4,5-四氢-2,8-二甲基-5-苯乙基-1H-吡啶并[4,3-b]吲哚(参见实施例47)的制备所述的相类似的方法,使用4-氯苯基肼盐酸盐、4-氟苯乙基溴和N-甲基-4-哌啶酮盐酸盐制备标题化合物。实施例67.2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(哌啶-1-基)乙酮的制备(化合物236)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸乙酯(200mg)和哌啶(2ml)的混合液在120℃加热8小时,经硅胶(230-400目)色谱用甲醇-二氯甲烷梯度洗脱而纯化,随后经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到5mg2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(哌啶-1-基)乙酮,为TFA盐。实施例68.2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(4-甲基哌啶-1-基)乙酮的制备(化合物237)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸乙酯(200mg)和4-甲基哌啶(2ml)的混合液在120℃加热8小时,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到40mg2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(4-甲基哌啶-1-基)乙酮。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例69.2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-吗啉代乙酮的制备(化合物238)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸乙酯(200mg)和吗啉(2ml)的混合液在120℃加热15小时,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到27mg2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-吗啉代乙酮。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例70.2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-硫代吗啉代乙酮的制备(化合物239)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸乙酯(200mg)和硫代吗啉(2ml)的混合液在120℃加热15小时,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到38mg2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-硫代吗啉代乙酮。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例71.3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(吡咯烷-1-基)丙-1-酮的制备(化合物240)将3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)丙酸乙酯(100mg)和吡咯烷(2ml)的混合液在120℃加热3小时,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到39mg3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(吡咯烷-1-基)丙-1-酮。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例72.1-(4-氨基哌啶-1-基)-3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙-1-酮的制备(化合物231)在25℃将1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-4-基氨基甲酸叔丁基酯(50mg)与三氟乙酸(2ml)在二氯甲烷(2ml)中搅拌16小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到10mg1-(4-氨基哌啶-1-基)-3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙-1-酮,为二-TFA盐。实施例76:化合物241的制备将Dimebon二盐酸盐(100mg)与SeO2(138mg)在吡啶(1mL)中混合,并将该反应混合液在80℃加热48小时。减压浓缩该反应混合液,并将得到的粗品产物经反相色谱纯化,得到28mg产物。实施例77:化合物189的制备依据通用方法8使用以下试剂:4-氯苯基肼、1-(2-氯乙基)哌啶盐酸盐、N-甲基-4-哌啶酮来制备标题化合物。实施例78:2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(吡咯烷-1-基)乙酮的制备(化合物243)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸乙酯(100mg)和吡咯烷(1ml)的混合液在120℃加热15小时,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到59mg2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(吡咯烷-1-基)乙酮。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例79:化合物244的制备使用以下试剂制备标题化合物:4-氯苯基肼盐酸盐、N-甲基-4-哌啶酮、溴乙酸乙酯、哌嗪。实施例80:化合物245的制备使用以下试剂制备标题化合物:4-氯苯基肼盐酸盐、N-甲基-4-哌啶酮、溴乙酸乙酯、2,6-二甲基哌嗪。实施例81:化合物246的制备使用以下试剂制备标题化合物:4-氯苯基肼盐酸盐、N-甲基-4-哌啶酮、溴乙酸乙酯、2-氧代哌嗪。实施例82:化合物247的制备依据通用方法8使用以下试剂:对-甲苯基肼盐酸盐、N-甲基-4-哌啶酮、4-甲氧基苯乙基溴来制备标题化合物。实施例83:化合物248的制备使用以下试剂制备标题化合物:4-氯苯基肼盐酸盐、N-甲基-4-哌啶酮、溴乙酸乙酯、1-异丙基哌嗪。实施例84:化合物249的制备依据通用方法8使用以下试剂:4-氟苯基肼盐酸盐、N-甲基-4-哌啶酮、4-氯苯乙基溴来制备标题化合物。实施例85:化合物250的制备依据通用方法1使用以下试剂:4-氯苯基肼盐酸盐、N-甲基-4-哌啶酮、2-(三氟甲基)-5-乙烯基吡啶来制备标题化合物。将2g5-溴-2-(三氟甲基)吡啶溶于DMF:THF(3:1,12mL)中。向该溶液中加入三丁基乙烯基锡(3g)和四(三苯基膦)钯(135mg)。将该反应混合液脱气,并用氮气净化5分钟,然后在100℃加热2小时,这时发现该反应完成(通过TLC监测)。用水稀释该反应混合液,并用乙酸乙酯萃取。经无水硫酸钠干燥合并的有机层,并减压蒸发(注意:产物可能是挥发性的),并将残余物经SiO2色谱(100-200目,洗脱:己烷-乙酸乙酯梯度洗脱)纯化。在40℃以下减压浓缩需要的级分,得到20.3g油状的2-(三氟甲基)-5-乙烯基吡啶。将222mg8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚溶于NMP(0.6mL)中。向该溶液中加入粉末状的KOH(196mg),并将该反应混合液在25℃搅拌10分钟。最后,加入2-(三氟甲基)-5-乙烯基吡啶(190mg),并将该反应混合液在45℃在密封管中加热30分钟。通过LCMS监测该反应。这段时间后,将该反应混合液冷却至25℃,并用饱和的NaCl水溶液(5mL)稀释。用乙酸乙酯萃取产物。经无水硫酸钠干燥合并的有机层,并减压蒸发,并将残余物经SiO2色谱(100-200目,洗脱:二氯甲烷-甲醇梯度)纯化。在40℃以下减压浓缩需要的级分,得到160mg产物。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例86:化合物251的制备使用以下试剂制备标题化合物:对-甲苯基肼盐酸盐、N-甲基-4-哌啶酮、溴乙酸乙酯、4-甲基哌啶。实施例87:化合物252的制备依据通用方法1使用以下试剂:对-甲苯基肼盐酸盐、N-甲基-4-哌啶酮、2-(三氟甲基)-5-乙烯基吡啶来制备标题化合物。将2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚二盐酸盐(203mg)溶于NMP(2.5mL)中。向该溶液中加入粉末状的KOH(463mg),并将该反应混合液在25℃搅拌10分钟。最后,加入2-(三氟甲基)-5-乙烯基吡啶(300mg),并将该反应混合液在25℃搅拌4小时。通过LCMS监测该反应。这段时间后,将该反应混合液冷却至25℃,并用饱和的NaCl水溶液(5mL)稀释。用乙酸乙酯萃取产物。经无水硫酸钠干燥合并的有机层,并减压蒸发,并将残余物经SiO2色谱(100-200目,洗脱:二氯甲烷-甲醇梯度)纯化。在40℃以下减压浓缩需要的级分,得到140mg产物。通过用无水THF中的草酸(1当量)处理将该游离碱转化为其草酸盐。实施例88:化合物253的制备依据通用方法8使用以下试剂:4-氟苯基肼盐酸盐、N-甲基-4-哌啶酮、4-氟苯乙基溴来制备标题化合物。实施例89:2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙-2-炔基)-1H-吡啶并[4,3-b]吲哚的制备将2,3,4,5-四氢-2,8-二甲基-5-(丙-2-炔基)-1H-吡啶并[4,3-b]吲哚(150mg,0.6mmol)、3-溴吡啶(0.6ml,0.6mmol)、二氯双(三苯基-膦)钯(8mg,0.012mmol)、CuI(1mg,0.006mmol)和三乙胺(0.01ml0.071mmol)在乙腈(5ml)中的混合液在80℃加热,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到108mg2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙-2-炔基)-1H-吡啶并[4,3-b]吲哚。2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙基)-1H-吡啶并[4,3-b]吲哚的制备将2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙-2-炔基)-1H-吡啶并[4,3-b]吲哚(20mg,0.06mmol)用10%Pd-C(10mg)在1个大气压的氢气下在甲醇(2ml)中氢化,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱而纯化后,得到5mg2,3,4,5-四氢-2,8-二甲基-5-(3-(吡啶-2-基)丙基)-1H-吡啶并[4,3-b]吲哚。通过用乙醇HCl处理将该游离碱转化为其HCl盐。实施例90:化合物255的制备将2,5-二溴吡啶(5g)溶于甲苯(30mL)中,并将该溶液冷却至-75℃。历经20分钟的时间向该溶液中滴加BuLi。将该反应物保持在-75℃达2小时。加入干冰(200g),并使该反应混合液温至25℃。将该反应混合液蒸发至干燥,将残余物溶于水(25mL)中,并通过加入浓HCl将pH调节至7。过滤固体产物,并用水洗涤,随后用乙醚洗涤。得到3g.TLCRf0.1,用50%乙酸乙酯-己烷。将5-溴吡啶-2-甲酸(10g)溶于260mLMeOH中。历经10分钟的时间向该溶液中滴加SOCl2(26mL)(注意:放热反应)。将该反应混合液加热至65℃达2小时,这时通过TLC发现该反应完成(将一等分试样蒸发至干燥,将残余物溶于饱和的NaHCO3水溶液中,并用乙酸乙酯萃取)。将该反应混合液蒸发至干燥。将残余物用饱和的NaHCO3水溶液碱化,并用乙酸乙酯萃取。将合并的乙酸乙酯层(Na2SO4)干燥,并减压蒸发,得到9.2g固体产物。TLCRf0.5(40%乙酸乙酯-己烷)。将4g5-溴吡啶-2-甲酸甲酯溶于160mL二烷中。在25℃向该溶液中加入三丁基乙烯基锡(11.7g),随后加入双(三苯基膦)二氯化钯(1.5g)。将该反应混合液脱气,并用氮气净化5分钟,然后在100℃加热2小时,这时发现该反应完成(通过TLC监测)。将该反应混合液减压蒸发(注意:产物可能是挥发性的),并将残余物经SiO2色谱(100-200目,洗脱:己烷-50%乙酸乙酯己烷梯度洗脱)纯化。在40℃以下减压浓缩需要的级分,得到2g产物,为浅黄色油状物(在-20℃为固体)。TLCRf0.3,使用40%EA-己烷。将2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚二盐酸盐(710mg)溶于中NMP(7.5mL)。向该溶液中加入粉末状的KOH(1.62g),并将该反应混合液在25℃搅拌10分钟。最后,历经5分钟滴加5-乙烯基吡啶-2-甲酸甲酯(977mg),并将该反应混合液在100℃在密封管中加热24小时。通过LCMS监测该反应。这段时间后,将该反应混合液冷却至25℃,并用水稀释(7mL)。通过加入甲酸将剩余溶液的pH调节为4-5。将得到的溶液直接经反相HPLC纯化。合并包含产物的需要的级分,并冻干(注意:化合物可能是热敏感的),得到370mg产物,为TFA盐,具有84%HPLC纯度(杂质是16%的未反应的咔啉)。通过第二次反相HPLC纯化纯度提高至98.5。实施例91:化合物256的制备将4-氨基苯甲酸甲酯(5.0g,0.0331mol)溶于浓HCl(50mL)中,并冷却至0℃。在0℃历经30分钟加入亚硝酸钠(2.5g,0.0331mol)的水溶液,并在室温搅拌90分钟。在0℃滴加在HCl(100mL)中的氯化锡(19.40g,0.0927mol),并在室温搅拌2小时。过滤反应混合液,并用乙醚洗涤得到的固体,并在旋转蒸发器干燥,得到6.2g92.53%收率的4-肼基苯甲酸甲酯,为白色固体。将4-肼基苯甲酸甲酯(1g,0.005mol)和5-(2-溴乙基)-2-甲基吡啶(1g,0.005mol)溶于三乙胺(2.1mL)中,并在室温搅拌1小时。将该反应在100℃加热,过夜。对反应物的LCMS分析显示形成产物。浓缩该反应混合液,向该反应混合液中加入水(20mL),并用乙酸乙酯混合液萃取。将粗品产物溶于乙酸乙酯中,并使用100%乙酸乙酯为洗脱剂,经中性氧化铝柱色谱纯化。色谱纯化后,得到400mg4-(1-(2-(6-甲基吡啶-3-基)乙基)肼基)苯甲酸甲酯,收率:28.5%。将4-(1-(2-(6-甲基吡啶-3-基)乙基)肼基)苯甲酸甲酯(0.1g,0.350mmol)和1-甲基哌啶-4-酮(0.052g,0.350mmol)溶于1MHCl(4mL)中,并在100℃加热18小时。该反应混合液的LCMS显示形成产物。浓缩该反应混合液,并经反相HPLC纯化,得到5mg2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-8-甲酸,为白色固体,收率:4%。实施例92:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(吡咯烷-1-基)乙酮的制备(化合物257)将2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙酸乙酯(100mg)和吡咯烷(1ml)的混合液在100℃加热15小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到60mg2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(吡咯烷-1-基)乙酮,为灰白色固体。实施例93:2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚2-氧化物的制备将Dimebon(500mg)溶于10mL二氯甲烷中。在25℃向其中滴加mCPBA(65%,536mg)在5mL二氯甲烷中的溶液。在25℃将该反应混合液搅拌24小时,这时发现该反应完成(通过TLC和LCMS)。减压除去溶剂(30℃以下),并将残余物经中性氧化铝柱色谱用甲醇-二氯甲烷梯度洗脱而纯化,得到220mg产物,为浅褐色油状物。TLCRf0.5,使用5%MeOH-DCM。该化合物还展现以下抑制特性:实施例94:3,4-二氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-甲酸2,2,2-三氯乙基酯的制备(化合物259)将1-(2-(6-甲基吡啶-3-基)乙基)-1-对-甲苯基肼(500mg)和N-三氯乙氧羰基-哌啶酮(560mg)在15mL乙醇HCl中混合,并搅拌20分钟。减压除去挥发物,将残余物溶于乙醇(5mL)中,并将该溶液在90℃加热3小时(通过LCMS和TLC监测该反应)。将该反应混合液冷却至25℃,并减压蒸发。将残余物用饱和的NaHCO3水溶液碱化,并用乙酸乙酯萃取。经Na2SO4干燥合并的乙酸乙酯层,并减压浓缩,得到粗品产物。将粗品产物经硅胶快速色谱(洗脱:己烷至10%丙酮己烷梯度洗脱)纯化,得到100mg产物,为淡黄色油状物。TLCRf0.2,使用20%丙酮-己烷。实施例95:2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物230)将3,4-二氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H吡啶并[4,3-b]吲哚2(5H)-甲酸2,2,2-三氯乙基酯(100mg)溶于冰醋酸(1.2mL)中和将该溶液在25℃搅拌10分钟。在25℃向该溶液中分批加入Zn粉(121mg),之后将该反应混合液在25℃搅拌过夜。将该反应混合液冷却至0℃,并用NH3水溶液将pH调节至8。将该反应混合液用乙酸乙酯萃取,经硫酸钠干燥合并的乙酸乙酯层,并浓缩,得到75mg产物,具有87%HPLC纯度。用在2mLTHF中的30mg草酸处理将产物(游离碱)转化为草酸盐。过滤得到的混悬液,得到产物。实施例96:2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-8-甲酸的制备(化合物261)将4-肼基苯甲酸甲酯(5.0g,0.0331mol)溶于50mL浓HCl中,并冷却至0℃。在0℃历经30分钟加入亚硝酸钠(2.5g,0.0331mol)水溶液,并在室温搅拌90分钟。在0℃滴加在100mLHCl中的氯化锡(19.40g,0.0927mol),并在室温搅拌2小时。过滤反应混合液,并用乙醚洗涤得到的固体,并在旋转蒸发器干燥,得到6.2g92.53%收率的4-肼基苯甲酸甲酯,为白色固体。将4-肼基苯甲酸甲酯(1g,0.005mol)和5-(2-溴乙基)-2-甲基吡啶(1g,0.005mol)溶于2.1mL三乙胺中,并在室温搅拌1小时。然后将反应在100℃加热,过夜。对该反应混合液的LCMS分析显示形成产物。将该反应混合液浓缩。向该反应混合液中加入水(20mL),并用乙酸乙酯萃取混合液。将粗品产物溶于乙酸乙酯中,并使用100%乙酸乙酯为洗脱剂,经中性氧化铝柱色谱纯化。色谱纯化后,得到400mg4-(1-(2-(6-甲基吡啶-3-基)乙基)肼基)苯甲酸甲酯,收率:28.5%。将4-(1-(2-(6-甲基吡啶-3-基)乙基)肼基)苯甲酸甲酯(0.1g,0.350mmol)和1-甲基哌啶-4-酮(0.052g,0.350mmol)溶于4mL1MHCl中,并在100℃加热18小时。该反应混合液的LCMS显示形成产物。浓缩该反应混合液,并经反相HPLC1纯化,得到5mg2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-8-甲酸,为白色固体,4%收率。实施例97:2,3,4,5-四氢-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-8-甲酸的制备(化合物274)将4-(1-(2-(6-甲基吡啶-3-基)乙基)肼基)苯甲酸甲酯盐酸盐(0.15mg,0.447mol)、哌啶酮(0.13mg,1.34mol)和7%硫酸在二烷(5mL)中的混合液在100℃加热2小时。冷却反应物质,并减压浓缩,得到粗品2,3,4,5-四氢-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-8-甲酸(LCMS;44%需要的峰),为褐色的油状物,将其经制备型HPLC纯化。实施例98:5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡啶-2-甲酸乙酯的制备(化合物275)将乙醇HCl加入化合物(2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙基)吡啶甲酸(0.750mg,2.14mmol)中,并回流7小时。浓缩该反应混合液,用饱和NaHCO3碱化,用乙酸乙酯(2x100mL)萃取,经硫酸钠干燥,并浓缩得到粗品产物。经硅胶(100-200)色谱(10%Me-OH在DCM中)纯化粗品产物,得到游离碱形式的5-(2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙基)吡啶甲酸乙酯。将乙醇HCl加入化合物5-(2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙基)吡啶甲酸乙酯(游离碱,0.07g,0.185mmol)中,然后使其保持一段时间,然后浓缩得到粘性化合物,将其用乙醚洗涤两至三次,得到5-(2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙基)吡啶甲酸乙酯盐酸盐。(50mg)。实施例99:2,3,4,5-四氢-2,8-二甲基-5-(2-(5-甲基噻吩-2-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物277)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基噻吩和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例100:4-(3,4-二氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-基)苯胺的制备(化合物278)由4-(3,4-二氢-8-甲基-1H-吡啶并[4,3-b]吲哚-2(5H)-基)苯胺、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例101:2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2-苯基-1H-吡啶并[4,3-b]吲哚的制备(化合物279)由2,3,4,5-四氢-8-甲基-2-苯基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例102:2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2-对-甲苯基-1H-吡啶并[4,3-b]吲哚的制备(化合物280)由2,3,4,5-四氢-8-甲基-2-对-甲苯基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例103:2-(4-溴苯基)-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物281)由2-(4-溴苯基)-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例104:2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2-(4-硝基苯基)-1H-吡啶并[4,3-b]吲哚的制备(化合物282)由2,3,4,5-四氢-8-甲基-2-(4-硝基苯基)-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例105:2-环丁基-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物283)由2-环丁基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例106:2,3,4,5-四氢-2-(4-甲氧基苯基)-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物284)由2,3,4,5-四氢-2-(4-甲氧基苯基)-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例107:2-(4-氟苯基)-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物285)由2-(4-氟苯基)-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例108:2-(4-氯苯基)-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物286)由2-(4-氯苯基)-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例109:2-(4-(二氟甲基)苯基)-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物287)由2-(4-(二氟甲基)苯基)-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例110:2-(4-(三氟甲基)苯基)-2,3,4,5-四氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物288)由2-(4-(三氟甲基)苯基)-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例111:2,3,4,5-四氢-2-(4-碘苯基)-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物289)由2,3,4,5-四氢-2-(4-碘苯基)-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例112:4-(3,4-二氢-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-2(5H)-基)苯酚的制备(化合物290)由4-(3,4-二氢-8-甲基-1H-吡啶并[4,3-b]吲哚-2(5H)-基)苯酚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例113:8-氯-7-氟-2,3,4,5-四氢-2-甲基-5-(2-(哌啶-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物292)根据通用方法8通过使用1-(溴甲基)哌啶、4-氯-3-氟苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例114:2,8-二甲基-5-(2-(6-甲基-1-氧化吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚2-氧化物的制备(化合物293)将2,3,4,5-四氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚(98mg,3.1mmol)与过氧化氢(30%在水中,0.2ml)在冰醋酸(1.2ml)中在865-70℃搅拌20小时,得到76mg相应的N,N'-二氧化物。实施例115:2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(2,2-二甲基哌啶-1-基)乙酮的制备(化合物294)根据通用方法8通过使用2-溴-1-(2,2-二甲基哌啶-1-基)乙酮、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例116:2-(8-氯-2-甲基-1,2,3,4-四氢-吡啶并[4,3-b]吲哚-5-基)-1-(1,1-二氧代-硫代吗啉-4-基)-乙酮的制备(化合物295)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸(100mg,0.35mmol)、硫代吗啉-1,1-二氧化物(48mg,0.35mmol)、DCC(81mg,0.39mmol)和DMAP(48mg,0.39mmol)在干燥的DCM(2.5mL)中的混合液在室温搅拌3小时。经硅藻土过滤该反应混合液,并通过旋转蒸发浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内10%B至80%B,进样体积:5mL)纯化后,得到10mg2-(8-氯-2-甲基-1,2,3,4-四氢-吡啶并[4,3-b]吲哚-5-基)-1-(1,1-二氧代-硫代吗啉-4-基)-乙酮,为TFA盐。实施例117:2-(8-氯-2-甲基-1,2,3,4-四氢-吡啶并[4,3-b]吲哚-5-基)-1-(1-氧代-硫代吗啉-4-基)-乙酮的制备(化合物296)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸(100mg,0.35mmol)、硫代吗啉-1,1-二氧化物(48mg,0.35mmol)、DCC(81mg,0.39mmol)和DMAP(48mg,0.39mmol)在干燥的DCM(2.5mL)中的混合液在室温搅拌3小时。经硅藻土过滤该反应混合液,并通过旋转蒸发浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内10%B至80%B,进样体积:5mL)纯化后,得到10mg2-(8-氯-2-甲基-1,2,3,4-四氢-吡啶并[4,3-b]吲哚-5-基)-1-(1-氧代-硫代吗啉-4-基)-乙酮,为TFA盐。实施例118:1-(4-乙酰基-哌嗪-1-基)-2-(8-氯-2-甲基-1,2,3,4-四氢-吡啶并[4,3-b]吲哚-5-基)-乙酮的制备(化合物297)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸(125mg,0.45mmol)和草酰氯(2ml)在二氯甲烷(6.0ml)中的混合液在25℃搅拌3小时。将该反应混合液浓缩至干燥。将得到的粗品溶于二氯甲烷(6.0ml)中,并加入DMAP(64mg,0.52mmol),随后加入乙酰基哌嗪(56mg,0.43mmol)。将得到的混合液在25℃搅拌14小时。在真空下除去溶剂,并经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到20mg1-(4-乙酰基-哌嗪-1-基)-2-(8-氯-2-甲基-1,2,3,4-四氢-吡啶并[4,3-b]吲哚-5-基)-乙酮,为三氟乙酸盐。实施例119:1-(3-氨基哌啶-1-基)-3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙-1-酮的制备(化合物298)根据通用方法8通过使用1-(3-氨基哌啶-1-基)-3-溴丙-1-酮、4-甲基苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例120:1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-3-甲酸的制备(化合物299)根据通用方法8通过使用1-(3-溴丙酰基)哌啶-3-甲酸、4-甲基苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例121:1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-4-甲酸的制备(化合物300)根据通用方法8通过使用1-(3-溴丙酰基)哌啶-4-甲酸、4-甲基苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例122:5-(4-氯苯乙基)-9-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物301)根据通用方法8通过使用1-(2-溴乙基)-4-氯苯、3-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例123:5-(4-氯苯乙基)-7-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物302)根据通用方法8通过使用1-(2-溴乙基)-4-氯苯、3-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例124:5-(4-氯苯乙基)-6-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物303)根据通用方法8通过使用1-(2-溴乙基)-4-氯苯、2-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例125:5-(4-氟苯乙基)-9-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物304)根据通用方法8通过使用1-(2-溴乙基)-4-氟苯、3-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例126:5-(4-氟苯乙基)-7-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物305)根据通用方法8通过使用1-(2-溴乙基)-4-氟苯、3-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例127:5-(4-氟苯乙基)-6-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物306)根据通用方法8通过使用1-(2-溴乙基)-4-氟苯、2-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例128:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(2-甲基吡咯烷-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物307)根据通用方法8通过使用1-(2-溴乙基)-2-甲基吡咯烷、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例129:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(3-甲基吡咯烷-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物308)根据通用方法8通过使用1-(2-溴乙基)-3-甲基吡咯烷、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例130:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(2,3-二甲基吡咯烷-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物309)根据通用方法8通过使用1-(2-溴乙基)-2,3-二甲基吡咯烷、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例131:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(2,2-二甲基吡咯烷-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物310)根据通用方法8通过使用1-(2-溴乙基)-2,2-二甲基吡咯烷、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例132:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(3,3-二甲基吡咯烷-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物311)根据通用方法8通过使用1-(2-溴乙基)-3,3-二甲基吡咯烷、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例133:8-氯-2,3,4,5-四氢-2-甲基-5-(3-(吡咯烷-1-基)丙基)-1H-吡啶并[4,3-b]吲哚(化合物312)的制备根据通用方法8通过使用1-(3-溴丙基)吡咯烷、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例134:1-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡咯烷-2-酮的制备(化合物313)根据通用方法8通过使用1-(2-溴乙基)吡咯烷-2-酮、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例135:1-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)哌啶-2-酮的制备(化合物314)根据通用方法8通过使用1-(2-溴乙基)哌啶-2-酮、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例136:1,2-二氢-1-羟基-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-4H-吡啶并[4,3-b]吲哚-3(5H)-酮的制备(化合物315)由1,2-二氢-1-羟基-2,8-二甲基-4H-吡啶并[4,3-b]吲哚-3(5H)-酮、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例137:3,4-二氢-4-羟基-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2H-吡啶并[4,3-b]吲哚-1(5H)-酮的制备(化合物317)由3,4-二氢-4-羟基-2,8-二甲基-2H-吡啶并[4,3-b]吲哚-1(5H)-酮、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例138:1,2-二氢-4-羟基-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-4H-吡啶并[4,3-b]吲哚-3(5H)-酮的制备(化合物318)由1,2-二氢-4-羟基-2,8-二甲基-4H-吡啶并[4,3-b]吲哚-3(5H)-酮、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例139:3,4-二氢-2-(羟基甲基)-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2H-吡啶并[4,3-b]吲哚-1(5H)-酮的制备(化合物319)由3,4-二氢-2-(羟基甲基)-8-甲基-2H-吡啶并[4,3-b]吲哚-1(5H)-酮、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例140:2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(2-甲基哌啶-1-基)乙酮的制备(化合物321)将2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酸(125mg,0.45mmol)和草酰氯(2ml)在二氯甲烷(6.0ml)中的混合液在25℃搅拌3小时。该反应完成后(通过TLC监测),将该反应混合液浓缩至干燥。向得到的粗品中加入二氯甲烷(6.0ml)和DMAP(64mg,0.52mmol),随后加入2-甲基哌啶(43mg,0.43mmol)。将得到的混合液在25℃搅拌14小时。该反应完成后(通过LCMS监测),在真空下除去溶剂,并经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到30mg2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(2-甲基哌啶-1-基)乙酮,为三氟乙酸盐。实施例141:2,8-二甲基-5-(2-(5-甲基吡嗪-2-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物322)向2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.5mmol)在N-甲基2-吡咯烷酮(0.75mL)中的溶液中加入粉末状的氢氧化钾(0.281g,5.0mmol),并使其在室温搅拌10分钟。加入2-甲基-5-乙烯基吡嗪(0.151g,1.2mmol),并在60℃再搅拌2小时。反应完成(TLC)后,将反应混合液用水(25mL)稀释,并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱纯化粗品(6%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸),得到所需化合物,为黄色油状物(0.05g,31%收率)。将2,8-二甲基-5-[2-(5-甲基-吡嗪-2-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.05g,0.156mmol)溶于THF(2.0mL)中。加入草酸二水合物(0.015g,0.000119mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟。过滤得到的沉淀物,并干燥,得到草酸盐,为白色固体(0.03g,47%收率)。实施例142:2,3,4,5-四氢-2,8-二甲基-5-(2-(2-甲基嘧啶-5-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物323)向2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.0005mol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.14g,0.0025mol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基嘧啶(0.151g,0.0012mol),并在80℃再搅拌3小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后经制备型TLC纯化,得到所需化合物,为黄色油状物(0.025g,收率16%)。将2,8-二甲基-5-[2-(2-甲基-嘧啶-5-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.025g,0.000078mol)溶于THF(1.0mL)中。加入草酸二水合物(0.0098g,0.000078mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.015g,收率47%)。实施例143:8-氯-5-(3-(6-(三氟甲基)吡啶-3-基)丙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物324)在冰冷的温度向氢化钠(0.014g,0.00054mol)在干燥的DMF(3mL)中的混悬液中,加入8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.00045mol),并在室温搅拌2小时。将该反应混合液冷却至-20℃,并加入5-(3-溴-丙基)-2-三氟甲基-吡啶(0.183g,0.00068mol),在15℃搅拌1小时,通过TLC监测反应。反应完成(TLC)后,将该反应混合液冷却至0℃,并加入水(10mL),用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(4%甲醇:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为褐色油状物(0.06g,收率32%)。将8-氯-2-甲基-5-[3-(6-三氟甲基-吡啶-3-基)-丙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.06g,0.000147mol)溶于THF(1.0mL)中,加入草酸二水合物(0.016g,0.000126mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为灰白色固体(0.050g,收率68%)。实施例144:3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(4-甲基哌啶-1-基)丙-1-酮的制备(化合物325)将3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)丙酸(50mg,0.17mmol)、4-甲基哌啶(0.02mL,0.17mmol)、DCC(38mg,0.18mmol)和DMAP(23mg,0.18mmol)在干燥的DCM(2mL)中的混合液在室温搅拌3小时。经硅藻土过滤该反应混合液,并浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内10%B至80%B,进样体积:5mL)纯化后,得到12mg3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(4-甲基哌啶-1-基)丙-1-酮,为TFA盐。实施例145:3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(哌啶-1-基)丙-1-酮的制备(化合物326)将3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)丙酸(0.1g,0.34mmol)溶于二氯甲烷(3mL)中,并将其冷却至0℃。向该反应混合液中滴加草酰氯(0.04mL,0.41mmol)。向该反应混合液中加入催化量(1滴)的二甲基甲酰胺。将该反应混合液在室温搅拌1小时。减压蒸馏掉过量的草酰氯。在室温、在氮气下向该残余物中加入哌啶(0.036mL,0.374mmol)在2mLDCM中的溶液和DMAP(0.055g,0.45mmol),并将反应物质在室温搅拌30分钟。将该反应混合液用水淬灭,并用10%NaHCO3中和,用乙酸乙酯(10mlx2)萃取。经硫酸钠干燥合并的有机层,并减压浓缩,得到粗品产物,将其经快速柱色谱使用甲醇:DCM(5:95)进一步纯化,得到12mg产物。将该产物在THF(2mL)和草酸(9mg,0.06mmol)中搅拌15分钟,并将混合液在真空下浓缩,得到11mg3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(哌啶-1-基)丙-1-酮,为草酸盐。实施例146:5-(4-氯苯乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚-8-甲酸的制备(化合物327)根据通用方法8通过使用1-(2-溴乙基)-4-氯苯、4-肼基苯甲酸甲酯、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例147:3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-吗啉代丙-1-酮的制备(化合物328)将3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)丙酸(100mg,0.34mmol)、吗啉(29mg,0.34mmol)、DCC(77mg,0.37mmol)和DMAP(46mg,0.37mmol)在干燥的DCM(2.5mL)中的混合液在室温搅拌3小时。经硅藻土过滤该反应混合液,并通过旋转蒸发器浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内10%B至80%B,进样体积:5mL)纯化后,得到10mg3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-吗啉代丙-1-酮,为TFA盐。实施例148:2,3,4,5-四氢-2-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物329)将2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(200mg,1mmol)、2-乙烯基吡啶(0.26mg,2.5mmol)、钠(5mg,0.21mmol)和CuSO4(5mg,催化的)在乙醇(4mL)在120℃加热16小时,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到60mg2,3,4,5-四氢-2-甲基-5-(2-(吡啶-2-基)乙基)-1H-吡啶并[4,3-b]吲哚,为三氟乙酸盐。实施例149:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-吗啉代乙酮的制备(化合物330)向2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙酸(100mg,0.38mmol)在DCM(10ml)中的溶液中加入DCC(95mg,0.46mmol)、DMAP(56mg,0.46mmol),并在室温搅拌5分钟,加入吗啉(1ml,11.5mmol),并在25℃搅拌14小时。将该反应混合液浓缩至干燥,并将得到的粗品经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到10mg(收率5.8%)2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-吗啉代乙酮,为TFA盐。实施例150:5-(3-(6-(三氟甲基)吡啶-3-基)丙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物331)在0℃向DMF(5mL)和氢化钠(0.024g,0.0006mol)的混悬液中加入2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.0005mol),并在室温搅拌2小时,将该反应混合液冷却至-20℃,并加入5-(3-溴-丙基)-2-三氟甲基-吡啶(0.2g,0.00075mol),在15℃搅拌1小时。反应完成(TLC)后,将反应物质冷却至0℃,并加入水(10mL),并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(3-4%甲醇:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为褐色油状物(0.04g,收率20%)。将2,8-二甲基-5-[3-(6-三氟甲基-吡啶-3-基)-丙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.04g,0.000103mol)溶于THF(1.0mL)中。加入草酸二水合物(0.012g,0.0000952mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为灰白色固体(0.030g,收率61%)。实施例151:2,3,4,5-四氢-2-甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚的制备(化合物332)将2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(186mg,1mmol)、5-(氯甲基)-2-甲基吡啶(324mg,2.3mmol)和60%NaH(120mg,3mmol)在DMF中(3ml)中的溶液在120℃加热16小时,经硅胶色谱(230-400目)使用甲醇-二氯甲烷梯度洗脱而纯化后,得到30mg2,3,4,5-四氢-2-甲基-5-((6-甲基吡啶-3-基)甲基)-1H-吡啶并[4,3-b]吲哚。实施例152:5-[2-(4-乙氧基-苯基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物333)将2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.2g,0.001mol)溶于N-甲基2-吡咯烷酮(3.0mL)中。加入粉末状的氢氧化钾(0.448g,0.008mol),并在120℃加热2小时。在相同的温度加入1-(2-溴-乙基)-4-乙氧基-苯(0.228g,0.001mol),并在室温搅拌3小时(TLC显示反应完成)。将该反应混合液用水(30mL)稀释,并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(4%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为淡黄色油状物(0.035g,收率10%)。将5-[2-(4-乙氧基-苯基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.035g,0.0001mol)溶于THF(0.5mL)中,加入草酸二水合物(0.013g,0.0001mol)在THF(0.5mL)中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为灰白色固体(0.015g,收率34%)。实施例153:5-[2-(4-乙氧基-苯基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物334)将2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.2g,0.001mol)溶于N-甲基2-吡咯烷酮(3.0mL)中,加入粉末状的氢氧化钾(0.448g,0.008mol),并在120℃加热2小时。在相同的温度加入1-(2-溴-乙基)-4-乙氧基-苯(0.228g,0.001mol),并在室温搅拌3小时(TLC显示反应完成)。将该反应混合液用水(30mL)稀释,并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(4%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为淡黄色油状物(0.035g,收率10%)。将5-[2-(4-乙氧基-苯基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.035g,0.0001mol)溶于THF(0.5mL)中,加入草酸二水合物(0.013g,0.0001mol)在THF(0.5mL)中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为灰白色固体(0.015g,收率34%)。实施例154:5-(4-叔-丁氧基苯乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物335)将2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.2g,0.001mol)溶于N-甲基2-吡咯烷酮(2.0mL)中。加入粉末状的氢氧化钾(0.5g,0.0089mol),并在100℃加热3小时。将反应物质冷却至室温,并在相同的温度加入1-(2-溴-乙基)-4-叔-丁氧基-苯(0.245g,0.001mol),并在室温搅拌2小时(TLC显示反应完成)。将该反应混合液用水(50mL)稀释,并用乙酸乙酯(2x100mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(0.5%MeOH:DCM,中性氧化铝,柱直径-2.5cm,氧化铝高度-大约5英寸)纯化粗品,然后经制备型TLC进一步纯化,得到所需化合物,为淡黄色油状物(0.024g,收率6%)。将5-[2-(4-叔-丁氧基-苯基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.024g,0.000063mol)溶于THF(1.0mL)中,加入草酸二水合物(0.008g,0.000063mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为灰白色固体(0.020g,收率67%)。实施例155:8-氯-5-[2-(4-甲氧基-苯基)-乙基]-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物336)将8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.22g,0.001mol)溶于N-甲基2-吡咯烷酮(3.0mL)中。加入粉末状的氢氧化钾(0.224g,0.004mol),并在100℃加热3小时。在相同的温度加入1-(2-溴-乙基)-4-甲氧基-苯(0.214g,0.001mol),并在室温搅拌6小时。将该反应混合液用水(30mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(4%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为淡黄色油状物(0.02g,收率6%)。将8-氯-5-[2-(4-甲氧基-苯基)-乙基]-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.020g,0.0564mol)溶于THF(1.0mL)中,加入草酸二水合物(0.007g,0.0564mol)在1.5mLTHF中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为白色固体(0.01g,收率40%)。实施例156:5-(3-氟-4-甲氧基苯乙基)-8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物337)将8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.5g,0.0022mol)溶于N-甲基2-吡咯烷酮(4.0mL)中。加入粉末状的氢氧化钾(1g,0.008mol),并在100℃加热2小时。在室温加入4-(2-溴-乙基)-2-氟-1-甲氧基-苯(0.520g,0.0022mol),并在室温搅拌0.5小时(TLC显示反应完成)。将该反应混合液用水(200mL)稀释,并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(4%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为淡黄色油状物(0.038g,收率4%)。将8-氯-5-[2-(3-氟-4-甲氧基-苯基)-乙基]-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.038g,0.00010mol)溶于THF(1.0mL)中。加入草酸二水合物(0.012g,0.00010mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为灰白色固体(0.016g,收率34%)。实施例157:8-氯-6-氟-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物338)向8-氯-6-氟-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(300mg,1.26mmol)在NMP(3.0ml)中的溶液中加入粉末状的KOH(705mg,12.57mmol),并在25℃搅拌10分钟。之后向上述的溶液中缓慢加入2-(三氟甲基)-5-乙烯基吡啶(435mg,2.5mmol),并在25℃搅拌24小时。该反应完成后(通过LCMS监测),向粗品中加入DM(去物质化)水,并用乙酸乙酯萃取。分离有机层,干燥,并浓缩。将得到的粗品经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到15mg8-氯-6-氟-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚,为TFA盐。实施例158:6,8-二氯-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物339)由6,8-二氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例159:7,9-二氟-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物340)由7,9-二氟-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例160:5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-8-(三氟甲氧基)-1H-吡啶并[4,3-b]吲哚的制备(化合物341)向5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-8-(三氟甲氧基)-1H-吡啶并[4,3-b]吲哚(200mg,0.74mmol)在NMP(1.5ml)中的溶液中,加入粉末状的KOH(415mg7.4mmol),并在25℃搅拌10分钟,之后向上述的溶液中缓慢加入2-(三氟甲基)-5-乙烯基吡啶(256mg,1.48mmol),并在25℃搅拌2小时。该反应完成后(通过LCMS监测),向粗品中加入DM水,并用乙酸乙酯萃取。干燥有机层,并浓缩。将得到的粗品经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到50mg(收率15.24%)5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-8-(三氟甲氧基)-1H-吡啶并[4,3-b]吲哚,为TFA盐。实施例161:8-异丙基-2-甲基-5-[2-(6-三氟甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物342)向8-异丙基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.43mmol)在N-甲基2-吡咯烷酮(0.5mL)中的溶液中加入粉末状的氢氧化钾(0.096g,1.72mmol),并将其在室温搅拌10分钟。加入2-三氟甲基-5-乙烯基吡啶(0.19g,1.1mmol),并在35℃再搅拌3小时。将该反应混合液用盐水(5mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱纯化粗品(5%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸),得到所需化合物,为褐色油状物(0.17g,收率45%)。将异丙基-2-甲基-5-[2-(6-三氟甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.080g,0.19mmol)溶于THF(2.0mL)中。加入草酸二水合物(0.017g,0.19mmol)在THF(2mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为白色固体(0.045g,收率46%)。实施例162:8-氯-7-氟-2-甲基-5-[2-(6-三氟甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物343)向8-氯-7-氟-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g.0.0041mol)在N-甲基2-吡咯烷酮(0.5mL)中的溶液中加入粉末状的氢氧化钾(0.08g,0.00143mol),并将其在室温搅拌10分钟。加入2-三氟甲基-5-乙烯基吡啶(0.079g,0.00046mol),并在室温再搅拌15小时,反应完成(TLC)后,将该反应混合液用水(5mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。将粗品经制备型HPLC纯化,得到所需化合物10,为淡黄色固体(0.02g,12%收率)。将8-氯-7-氟-2-甲基-5-[2-(6-三氟甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.02g,0.0000485mol)溶于THF(1.0mL)中。加入草酸二水合物(0.006g,0.0000485mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为白色固体(0.015g,收率62%)。实施例163:8-氯-9-氟-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物344)由8-氯-9-氟-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例164:7-氮杂-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物345)由9-氮杂-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例165:9-氮杂-2-甲基-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物346)由9-氮杂-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例166:8-氮杂-2-甲基-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物347)由8-氮杂-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例167:6,8-二氮杂-2-甲基-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物348)由6,8-二氮杂-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例168:8-氯-2-甲基-5-(2-吡咯-1-基-乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物349)将8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.45mmol)溶于N-甲基2-吡咯烷酮(3.0mL)中。加入粉末状的氢氧化钾(0.224g,4mmol),并在100℃加热3小时。在相同的温度加入2-(2-溴-乙基)吡啶(0.09g,0.51mol),并在室温搅拌3小时。将该反应混合液用水(30mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(4%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为淡黄色油状物(0.01g,收率7%)。将8-氯-2-甲基-5-(2-吡咯-1-基-乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.010g,0.0319mmol)溶于THF(0.5mL)中,加入草酸二水合物(0.004g,0.0319mol)在THF(0.5mL)中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为白色固体(0.01g,收率78%)。实施例169:8-氯-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1,2-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物350)由8-氯-2,3,4,5-四氢-1,2-二甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例170:3-(8-氯-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-基)环戊醇的制备(化合物351)将3-(8-氯-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-基)环戊醇(300mg,1.03mmol)、2-三氟甲基-5-乙烯基吡啶(140mg,0.809mmol)和氢氧化钾(310mg,5.5mmol)在0.6mlNMP中加热至60℃达2小时。将反应物质在室温冷却,并用20ml乙酸乙酯稀释。用盐水洗涤有机层,并经硫酸钠干燥,并在真空下浓缩。将其经用三乙胺去活化的230-400目硅胶(快速色谱)使用乙酸乙酯/甲醇(5-10%)作为洗脱液来纯化,随后经制备型HPLC分离3-(8-氯-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-基)环戊醇。获得70mgTFA盐。实施例171:8-氯-4-氟-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物352)由8-氯-4-氟-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例172:9-氮杂-2,8-二甲基-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物353)将咔啉(150mg,0.746mmol,1当量)、2-三氟甲基-5-乙烯基吡啶(142mg,0.82mmol,1.1当量)和KOH(146mg,2.61mmol,3.5当量)与0.45mlNMP在50℃搅拌3.5小时。然后将该反应混合液用乙酸乙酯(15ml)稀释,并用盐水洗涤。将有机部分在真空下蒸发,并经中性氧化铝柱使用乙酸乙酯/MeOH(0-100%)梯度洗脱而纯化。使用制备型HPLC(LC-MS)进一步纯化含所需物质的级分。获得35mgTFA盐。实施例173:8-氯-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2,3-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物354)由8-氯-2,3,4,5-四氢-2,3-二甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例174:6-氮杂-2,8-二甲基-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物355)由6-氮杂-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例175:7-氮杂-2,8-二甲基-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物356)由7-氮杂-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例176:8-乙基-2-甲基-5-[2-(6-甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物357)向8-乙基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.107g,0.5mmol)在N-甲基2-吡咯烷酮(2mL)中的溶液中加入粉末状的氢氧化钾(0.224g,4.0mmol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基吡啶(0.065g,0.55mmol),并在120℃再搅拌24小时。通过TLC监测反应完成后,将该反应混合液用水(30mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱(8%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为褐色油状物(0.034g,收率20%)。将8-乙基-2-甲基-5-[2-(6-甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.034g,0.102mmol)溶于THF(0.5mL)中。加入草酸二水合物(0.013g,0.102mol)在THF(0.5mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.015g,收率32%)。实施例177:8-乙基-2-甲基-5-[2-(6-三氟甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物358)向8-乙基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.107g.0.0005mol)在N-甲基2-吡咯烷酮(2.0mL)中的溶液中加入粉末状的氢氧化钾(0.224g,0.004mol),并将其在室温搅拌10分钟。加入2-三氟甲基-5-乙烯基吡啶(0.095g,0.00055mol),并在60℃再搅拌1小时,通过TLC监测反应完成后,将该反应混合液用盐水(5mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(4%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为褐色油状物(0.11g,收率57%)。将8-乙基-2-甲基-5-[2-(6-三氟甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.000258mol)溶于THF(2.0mL)中。加入草酸二水合物(0.032g,0.000258mol)在THF(2mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为白色固体(0.080g,收率65%)。实施例178:5-[2-(6-乙基-吡啶-3-基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物359)向2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.15g.0.749mmol)在N-甲基2-吡咯烷酮(1.2mL)中的溶液中加入粉末状的氢氧化钾(0.42g,7.49mmol),并将其在室温搅拌10分钟。加入2-乙基-5-乙烯基-吡啶(0.299g,2.24mmol),并在100℃再搅拌18小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径2.5cm,硅胶高度-大约5英寸)纯化粗品,然后进一步经制备型TLC纯化,得到所需化合物,为黄色油状物(0.045g,收率18%)。将5-[2-(6-乙基-吡啶-3-基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.040g,0.12mol)溶于THF(1.0mL)中。加入草酸二水合物(0.012g,0.096mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.030g,收率60%)。实施例179:5-[2-(6-异丙基-吡啶-3-基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物360)向2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.5mmol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.140g,2.5mmol),并将其在室温搅拌10分钟。加入2-异丙基-5-乙烯基-吡啶(0.185g,1.25mol),并在100℃再搅拌4小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后进一步经制备型TLC纯化,得到所需化合物,为黄色油状物(0.025g,收率14%)。将5-[2-(6-异丙基-吡啶-3-基)-乙基]-2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.025g,0.00007mol)溶于THF(1.0mL)中。加入草酸二水合物(0.0095g,0.00007mol)在THF(0.5mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.024g,收率77%)。实施例180:8-氯-2-甲基-5-[2-(2-甲基-嘧啶-5-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物361)向8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.075g,0.34mmol)在N-甲基2-吡咯烷酮(0.5mL)中的溶液中加入粉末状的氢氧化钾(0.095g,1.7mmol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基嘧啶(0.102g,0.85mmol),并在80℃再搅拌3小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后经制备型TLC纯化,得到所需化合物,为黄色油状物(0.020g,收率13%)。将8-氯-2-甲基-5-[2-(2-甲基-嘧啶-5-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.020g,0.06mmol)溶于THF(1.0mL)中。加入草酸二水合物(0.008g,0.06mmol)在THF(0.5mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.008g,收率32%)。实施例181:8-乙基-2-甲基-5-[2-(2-甲基-嘧啶-5-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物362)向8-乙基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.00046mol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.13g,0.0023mol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基嘧啶(0.14g,0.00116mol),并在80℃再搅拌3小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后经制备型TLC纯化,得到所需化合物。为黄色油状物(0.032g,收率20%)。将8-乙基-2-甲基-5-[2-(2-甲基-嘧啶-5-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.032g,0.000095mol)溶于THF(1.0mL)中。加入草酸二水合物(0.012g,0.000095mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.030g,收率75%)。实施例182:8-氯-2-甲基-5-[2-(5-甲基-吡嗪-2-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物363)向8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.11g,5.0mmol)在N-甲基2-吡咯烷酮(2.0mL)中的溶液中加入粉末状的氢氧化钾(0.224g,4.0mmol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基吡嗪(0.065g,0.55mmol),并在60℃再搅拌2小时。反应完成(TLC)后,将反应混合液用水(20mL)稀释,并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱纯化粗品(6%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸),得到所需化合物,为黄色油状物(0.09g,收率54%)。8-氯-2-甲基-5-[2-(5-甲基-吡嗪-2-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.09g,0.26mmol)将溶于THF(1.0mL)中。加入草酸二水合物(0.034g,0.26mmol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为白色固体(0.08g,收率68%)。实施例183:8-乙基-2-甲基-5-[2-(5-甲基-吡嗪-2-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物364)向8-乙基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.054g.0.25mmol)在N-甲基2-吡咯烷酮(2.0mL)中的溶液中加入粉末状的氢氧化钾(0.112g,0.002mol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基吡嗪(0.033g,0.75mmol),并在60℃再搅拌1小时。反应完成(TLC)后,将反应混合液用水(20mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,得到所需化合物,为黄色油状物(0.05g,收率60%)。将8-乙基-2-甲基-5-[2-(5-甲基-吡嗪-2-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.05g,0.000149mol)溶于THF(1.0mL)中。加入草酸二水合物(0.019g,0.000149mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.035g,收率50%)。实施例184:8-氯-5-(2-(4-(三氟甲基)-6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物365)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、4-(三氟甲基)-2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例185:8-乙基-5-(2-(4-(三氟甲基)-6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物366)由8-乙基-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、4-(三氟甲基)-2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例186:5-(2-(4-(三氟甲基)-6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物367)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、4-(三氟甲基)-2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例187:5-(2-(4-(三氟甲基)-6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-8-异丙基-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物368)由2,3,4,5-四氢-8-异丙基-2-甲基-1H-吡啶并[4,3-b]吲哚、4-(三氟甲基)-2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例188:8-异丙基-2-甲基-5-[2-(5-甲基-吡嗪-2-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物369)向8-异丙基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.2g,0.87mmol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.5g,8.7mmol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基吡嗪(0.25g,2.1mmol),并在60℃再搅拌5小时。反应完成(TLC)后,将反应混合液用水(20mL)稀释,并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(6%MeOH:DCM,硅胶100-200目,柱直径2.5cm,硅胶高度-大约5英寸)纯化粗品,然后进一步经制备型TLC纯化,得到所需化合物,为黄色固体(0.05g,收率16%)。将8-异丙基-2-甲基-5-[2-(5-甲基-吡嗪-2-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.05g,0.143mmol)溶于THF(2.0mL)中。加入草酸二水合物(0.018g,0.143mmol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.02g,收率32%)。实施例189:8-异丙基-2-甲基-5-[2-(2-甲基-嘧啶-5-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物370)向8-异丙基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.000438mol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.122g,0.00021mol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基嘧啶(0.13g,0.00109mol),并在80℃再搅拌3小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后经制备型TLC纯化。得到所需化合物,为黄色油状物(0.036g,收率24%)。将8-异丙基-2-甲基-5-[2-(2-甲基-嘧啶-5-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.036g,0.000103mol)溶于THF(1.0mL)中。加入草酸二水合物(0.013g,0.000103mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.022g,收率49%)。实施例190:8-氯-5[2-(6-乙基-吡啶-3-基)-乙基]-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物371)向8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.15g,0.68mmol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.4g,6.8mmol),并将其在室温搅拌10分钟。加入2-乙基-5-乙烯基-吡啶(0.28g,2.04mmol),并在100℃再搅拌18小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后进一步经制备型TLC纯化,得到所需化合物,为黄色油状物(0.025g,收率10%)。将8-氯-5-[2-(6-乙基-吡啶-3-基)-乙基]-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.025g,0.07mmol)溶于THF(1.0mL)中。加入草酸二水合物(0.009g,0.07mmol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.010g,收率32%)。实施例191:8-氯-5-[2-(6-异丙基-吡啶-3-基)-乙基]-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物372)向8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.00045mol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.140g,0.0025mol),并将其在室温搅拌10分钟。加入2-异丙基-5-乙烯基-吡啶(0.185g,0.00125mol),并在100℃再搅拌4小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后进一步经制备型TLC纯化,得到所需化合物,为黄色油状物(0.027g,收率15%)。将8-氯-5-[2-(6-异丙基-吡啶-3-基)-乙基]-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.027g,0.00007mol)溶于THF(1.0mL)中。加入草酸二水合物(0.009g,0.00007mol)在THF(0.5mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.027g,收率81%)。实施例192:8-氯-5-(2-(2-(三氟甲基)嘧啶-5-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物373)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基嘧啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例193:8-氯-5-(2-(5-(三氟甲基)吡嗪-2-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物374)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基嘧啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例194:2',8'-二甲基-5'-(2-(6-(三氟甲基)吡啶-3-基)乙基)-1',2',4',5'-四氢螺[环丙烷-1,3'-吡啶并[4,3-b]吲哚]的制备(化合物375)由2',8'-二甲基-1',2',4',5'-四氢螺[环丙烷-1,3'-吡啶并[4,3-b]吲哚]、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例195:2',8'-二甲基-5'-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2',3',4',5'-四氢螺[环丙烷-1,1'-吡啶并[4,3-b]吲哚]的制备(化合物376)由2',8'-二甲基-2',3',4',5'-四氢螺[环丙烷-1,1'-吡啶并[4,3-b]吲哚]、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例196:5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2,3,3,8-四甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物377)由2,3,4,5-四氢-2,3,3,8-四甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例197:5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1,1,2,8-四甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物378)由2,3,4,5-四氢-1,1,2,8-四甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例198:2,3,4,5-四氢-2,6-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物379)由2,3,4,5-四氢-2,6-二甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例199:5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2,6-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物380)向2,3,4,5-四氢-2,6-二甲基-1H-吡啶并[4,3-b]吲哚(200mg,1.0mmol)在NMP(2.5ml)中的溶液中加入粉末状的KOH(561mg,10mmol),并在25℃搅拌10分钟,之后向上述的溶液中缓慢加入2-(三氟甲基)-5-乙烯基吡啶(346mg,2.0mmol),并在25℃搅拌16小时。该反应完成后(通过LCMS监测),向该粗品中加入水(5mL),并用乙酸乙酯萃取。干燥有机层,并浓缩。将得到的粗品经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到40mg(8.2%)5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2,6-二甲基-1H-吡啶并[4,3-b]吲哚,为TFA盐。实施例200:3-(6-(三氟甲基)吡啶-3-基)-2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸的制备(化合物381)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、(Z)-3-(6-(三氟甲基)吡啶-3-基)丙烯酸和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例201:3-(6-(三氟甲基)吡啶-3-基)-2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸乙酯的制备(化合物382)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、(Z)-3-(6-(三氟甲基)吡啶-3-基)丙烯酸乙基酯和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例202:2-(6-(三氟甲基)吡啶-3-基)-3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸的制备(化合物383)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-(6-(三氟甲基)吡啶-3-基)丙烯酸和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例203:2-(6-(三氟甲基)吡啶-3-基)-3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸甲酯的制备(化合物384)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-(6-(三氟甲基)吡啶-3-基)丙烯酸甲酯和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例204:5-(2-(2-(三氟甲基)嘧啶-5-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物385)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基嘧啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例205:5-(2-(5-(三氟甲基)吡嗪-2-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物386)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡嗪和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例206:2,8-二甲基-5-(2-嘧啶-5-基-乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物387)向2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.49mmol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.14g,2.5mmol),并将其在室温搅拌10分钟。加入5-乙烯基嘧啶(0.132g,1.25mmol),并在60℃再搅拌5小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。将粗品经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度大约5英寸)纯化,然后进一步经制备型TLC纯化,得到所需化合物,为黄色油状物(0.050g,收率32.6%)。将2,8-二甲基-5-(2-嘧啶-5-基-乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.040g,0.13mol)溶于THF(1.0mL)中。加入草酸二水合物(0.015g,0.119mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.046g,收率90%)。实施例207:8-氯-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2,3,3-三甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物388)由8-氯-2,3,4,5-四氢-2,3,3-三甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例208:8-氯-5-(2-(6-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1,1,2-三甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物389)由8-氯-2,3,4,5-四氢-1,1,2-三甲基-1H-吡啶并[4,3-b]吲哚、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例209:8'-氯-2'-甲基-5'-(2-(6-(三氟甲基)吡啶-3-基)乙基)-1',2',4',5'-四氢螺[环丙烷-1,1'-吡啶并[4,3-b]吲哚]的制备(化合物390)由8'-氯-2'-甲基-2',3',4',5'-四氢螺[环丙烷-1,1'-吡啶并[4,3-b]吲哚]、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例210:8'-氯-2'-甲基-5'-(2-(6-(三氟甲基)吡啶-3-基)乙基)-1',2',4',5'-四氢螺[环丙烷-1,3'-吡啶并[4,3-b]吲哚]的制备(化合物391)由8'-氯-2'-甲基-1’,2',4',5'-四氢螺[环丙烷-1,3'-吡啶并[4,3-b]吲哚]、2-(三氟甲基)-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例211:8-异丙基-2-甲基-5-[2-(6-甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物392)向8-异丙基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.2g,0.877mmol)在N-甲基2-吡咯烷酮(2mL)中的溶液中加入粉末状的氢氧化钾(0.2g,3.5mmol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基吡啶(0.104g,0.877mmol),并在120℃再搅拌20小时。将该反应混合液用水(30mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱(5%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,并进一步用制备型TLC纯化,得到所需化合物,为褐色油状物(0.045g,收率15%)。将8-异丙基-2-甲基-5-[2-(6-甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.045g,0.129mmol)溶于THF(2.0mL)中。加入草酸二水合物(0.016g,0.129mmol)在THF(2mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.045g,收率80%)。实施例212:1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-4-基氨基甲酸叔丁基酯的制备(化合物393)将3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酸(500mg,1.8mmol)与哌啶-4-基氨基甲酸叔丁基酯(0.366ml,1.8mmol)、EDCI-HCl(0.35g,1.8mmol)和三乙胺(0.253ml,1.8mmol)在二氯甲烷(20ml)中搅拌,经中性氧化铝色谱用甲醇-二氯甲烷梯度洗脱纯化,随后经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到50mg1-(3-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙酰基)哌啶-4-基氨基甲酸叔丁基酯,为三氟乙酸盐。实施例213:5-(2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙基)-2-甲基吡啶1-氧化物的制备(化合物394)向在乙酸(0.3ml)和甲醇(2ml)中的N,N'-二氧化物中间体(76mg,0.21mmol)中加入亚硫酸氢钠(40%在水中,0.2ml),并将该反应混合液在0℃搅拌0.5小时。将该反应混合液用饱和的NaHCO3水溶液碱化,并用乙酸乙酯萃取,得到50mg2,3,4,5-四氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚N'-氧化物。实施例214:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(哌啶-1-基)乙酮的制备(化合物395)将2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙酸(1.5g,5.8mmol)溶于二氯甲烷(15mL)中,并使用冰浴将其冷却至0℃;滴加草酰氯(0.61mL,6.9mmol),向该反应混合液中加入催化量(2滴)的二甲基甲酰胺。加入后,将该反应混合液在室温搅拌1小时。减压蒸馏掉过量的草酰氯。在室温、在氮气下向该残余物中加入哌啶(0.68mL,6.9mmol)在4mLDCM中的溶液和DMAP(0.71g,5.8mmol),并将反应物质在室温搅拌0.5小时。通过冰水淬灭该反应混合液,并用二氯甲烷萃取,并经柱色谱纯化,得到0.8g所需游离碱形式的化合物。通过乙醇HCl处理将该游离碱转化为HCl盐。用己烷冲洗氢化钠(0.8g,20mmol)来除去油,并将其在真空下干燥。然后将氢化钠溶于THF中。在0℃向该溶液中滴加在THF中的2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(2g,10mmol)。然后将该反应混合液搅拌0.5小时。将2-氯-1-(哌啶-1-基)乙酮(1.9g,12mmol)在THF中的溶液滴加入反应混合液中。然后将反应混合液在室温搅拌2小时。通过TLC监测反应。反应完成后,用冰水淬灭反应混合液。将THF蒸发,并用乙酸乙酯萃取水层。经无水硫酸钠干燥有机层。用己烷和乙醚洗涤粗品化合物以除去有颜色的杂质,然后通过使用甲醇重结晶,得到1g所需化合物,然后将其与乙醇HCl搅拌,得到2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)-1-(哌啶-1-基)乙酮的HCl盐。实施例215:8-叔-丁基-2,3,4,5-四氢-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物396)向8-叔-丁基-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(200mg,0.82mmol)在NMP(3.0ml)中的溶液中加入粉末状的KOH(462mg,8.23mmol),并在25℃搅拌10分钟,之后向上述的溶液中缓慢加入2-(三氟甲基)-5-乙烯基吡啶(286mg,1.65mmol),并在25℃搅拌14小时。该反应完成后(通过LCMS监测),向该粗品中加入水(5mL),并用乙酸乙酯萃取。干燥有机层,并浓缩。将得到的粗品经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到30mg8-叔-丁基-2,3,4,5-四氢-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚,为TFA盐。实施例216:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(异二氢吲哚-2-基)乙酮的制备(化合物397)将2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙酸(50mg,0.205mmol)、异二氢吲哚(23mg,0.205mmol)、DCC(46mg,0.22mmol)和DMAP(27mg,0.22mmol)在干燥的DCM(2.0mL)中的混合液在室温搅拌3小时。经硅藻土过滤该反应混合液,并使用旋转蒸发器浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后得到16.3mg2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(异二氢吲哚-2-基)乙酮,为TFA盐。实施例217:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(3,4-二氢异喹啉-2(1H)-基)乙酮的制备(化合物398)将2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙酸(50mg,0.205mmol)、1,2,3,4-四氢异喹啉(25mg,0.205mmol)、DCC(46mg,0.22mmol)和DMAP(27mg,0.22mmol)在干燥的DCM(2.0mL)中的混合液在室温搅拌3小时。经硅藻土过滤该反应混合液,并使用旋转蒸发器浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到7.9mg2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(3,4-二氢异喹啉-2(1H)-基)乙酮,为TFA盐。实施例218:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-硫代吗啉代乙酮的制备(化合物399)将2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙酸(50mg,0.205mmol)、硫代吗啉(19mg,0.205mmol)、DCC(46mg,0.22mmol)和DMAP(27mg,0.22mmol)在干燥的DCM(2.0mL)中的混合液在室温搅拌3小时。经硅藻土过滤该反应混合液,并使用旋转蒸发器浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到34.2mg2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-硫代吗啉代乙酮,为TFA盐。实施例219:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(2-(吡啶-3-基)吡咯烷-1-基)乙酮的制备(化合物400)根据通用方法8通过使用2-溴-1-(2-(吡啶-3-基)吡咯烷-1-基)乙酮、(2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚)、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例220:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(3-甲基哌啶-1-基)乙酮的制备(化合物401)将2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙酸(50mg,0.205mmol)、3-甲基哌啶(19mg,0.205mmol)、DCC(46mg,0.22mmol)和DMAP(27mg,0.22mmol)在干燥的DCM(2.0mL)中的混合液在室温搅拌3小时。经硅藻土过滤该反应混合液,并使用旋转蒸发器浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到34.2mg2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(3-甲基哌啶-1-基)乙酮,为TFA盐。实施例221:2,3,4,5-四氢-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-8-甲酸的制备(化合物402)向2,3,4,5-四氢-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-8-甲酸(1.0g,4.09mmol)在NMP(7.8ml)中的溶液中加入粉末状的KOH(2.3g,40.9mmol),并在25℃搅拌10分钟。之后向上述的溶液中缓慢加入2-(三氟甲基)-5-乙烯基吡啶(1.4g,8.1mmol),并在45℃搅拌14小时。反应完成后(通过LCMS监测),向粗品中加入DM水,并用乙酸乙酯萃取。分离有机层,干燥,并浓缩。将得到的粗品经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化,得到15mg2,3,4,5-四氢-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚-8-甲酸,为TFA盐。实施例222:8-氯-5-[2-(6-甲基-吡啶-3-基)-乙基]-2-(3-三氟甲基-苯基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物403)在室温向N-(4-氯-苯基)-N-[2-(6-甲基-吡啶-3-基)-乙基]-肼盐酸盐(0.13g,0.5mmol)在无水乙醇(5mL)中的搅拌着的溶液中加入1-(3-三氟甲基-苯基)-哌啶-4-酮(0.122g,0.5mmol),然后回流1小时。反应完成(TLC)后,将乙醇蒸发。将粗品经制备型HPLC纯化,得到所需化合物,为褐色固体(0.02g,收率10%)。将8-氯-5-[2-(6-甲基-吡啶-3-基)-乙基]-2-(3-三氟甲基-苯基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.020g,0.0425mmol)溶于THF(1.0mL)中。加入草酸二水合物(0.0053g,0.0425mmol)在1.5mLTHF中的溶液,并在室温搅拌30分钟。过滤沉淀并干燥,得到草酸盐,为褐色固体(0.01g,收率43%)。实施例223:8-氯-7-氟-2-甲基-5-[2-(6-甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物404)向8-氯-7-氟-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.3g,1.25mmol)在N-甲基2-吡咯烷酮(1.5mL)中的溶液中加入粉末状的氢氧化钾(0.7g,12.5mmol),并将其在室温搅拌10分钟。加入2-三氟甲基-5-乙烯基吡啶(0.44g,3.7mmol),并在120℃再搅拌24小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x150mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。将粗品经柱色谱(6%甲醇:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化,然后进一步经制备型HPLC纯化,得到所需化合物10,为淡黄色固体(0.080g,收率21%)。将8-氯-7-氟-2-甲基-5-[2-(6-甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.08g,0.22mmol)溶于THF(2.0mL)中。加入草酸二水合物(0.028g,0.22mmol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为白色固体(0.070g,收率70%)。实施例224:5-(2-(6-环丙基吡啶-3-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物405)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-环丙基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例225:8-氯-5-(2-(6-环丙基吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物406)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-环丙基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例226:2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(6-(三氟甲基)吡啶-3-基)乙醇的制备(化合物407)将氢化钠(38mg,1.6mmol,1.2当量)加入8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF中(6ml)中的溶液中,并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(4-氟苯基)-2-甲基环氧乙烷(400mg,2.1mmol,1.6当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例227:5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡啶-2(1H)-酮的制备(化合物408)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、5-乙烯基吡啶-2(1H)-酮和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例228:5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡啶-2(1H)-酮的制备(化合物409)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、5-乙烯基吡啶-2(1H)-酮和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例229:5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)-1-甲基吡啶-2(1H)-酮的制备(化合物410)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、1-甲基-5-乙烯基吡啶-2(1H)-酮和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例230:1-环丙基-5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡啶-2(1H)-酮的制备(化合物411)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚,1-环丙基-5-乙烯基吡啶-2(1H)-酮和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例231:5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)-1-甲基吡啶-2(1H)-酮的制备(化合物412)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、1-甲基-5-乙烯基吡啶-2(1H)-酮和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例232:5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)-1-环丙基吡啶-2(1H)-酮的制备(化合物413)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、1-环丙基-5-乙烯基吡啶-2(1H)-酮和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例233:1-(三氟甲基)-5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡啶-2(1H)-酮的制备(化合物414)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、1-(三氟甲基)-5-乙烯基吡啶-2(1H)-酮和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例234:5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)-1-(三氟甲基)吡啶-2(1H)-酮的制备(化合物415)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、1-(三氟甲基)-5-乙烯基吡啶-2(1H)-酮和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例235:8-氯-2-(4-氟苯基)-2,3,4,5-四氢-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物416)由8-氯-2-(4-氟苯基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例236:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(4-甲基哌嗪-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物417)根据通用方法8通过使用1-(2-溴乙基)-4-甲基哌嗪、4-氯苯基肼盐酸盐、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例237:2,3,4,5-四氢-2,8-二甲基-5-(2-(4-甲基哌嗪-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物418)将LAH(0.022g,0.58mmol)加入干燥的THF(3mL),并冷却至0℃,将2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)-1-(4-甲基哌嗪-1-基)乙酮(0.05g,0.147mmol)分批加入其中。将该反应混合液回流4小时。将该反应混合液冷却至0℃,并用饱和的Na2SO4淬灭。经硅藻土过滤形成的固体,用THF洗涤,经Na2SO4干燥,并减压浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到11mg2,8-二甲基-5-(2-(4-甲基哌嗪-1-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚,为TFA盐。实施例238:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(1-甲基异喹啉-4-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物420)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、1-甲基-4-乙烯基异喹啉和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例239:2,3,4,5-四氢-2,8-二甲基-5-(2-(1-甲基异喹啉-4-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物421)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、1-甲基-4-乙烯基异喹啉和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例240:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(喹啉-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物422)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、3-乙烯基喹啉和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例241:2,3,4,5-四氢-2,8-二甲基-5-(2-(喹啉-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物423)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、3-乙烯基喹啉和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例242:8-氯-2-甲基-5-(2-嘧啶-5-基-乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物424)向8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.15g,0.68mmol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.19g,3.4mmol),并将其在室温搅拌10分钟。加入5-乙烯基嘧啶(0.177g,1.7mmol),并在60℃再搅拌5小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x100mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后进一步经制备型TLC纯化,得到所需化合物,为黄色油状物(0.030g,收率14%)。将8-氯-2-甲基-5-(2-嘧啶-5-基-乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.025g,0.076mmol)溶于THF(1.0mL)中。加入草酸二水合物(0.0096g,0.076mol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.025g,收率78%)。实施例243:5-(2-(5-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物425)将2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.15g,0.75mmol)、3-(三氟甲基)-5-乙烯基吡啶(0.13g,0.75mmol)和四丁基氯化铵(2mg,0.0375mmol)加入5ml50%NaOH水溶液中。将该反应混合液在100℃加热8小时。将反应物质在室温冷却,加入5ml水,并用乙酸乙酯萃取。经无水硫酸钠干燥有机层,浓缩,并经制备型HPLC纯化,得到2,8-二甲基-5-(2-(5-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(35mg)。实施例244:8-氯-5-(2-(5-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物426)将8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.165g,0.75mmol)、3-(三氟甲基)-5-乙烯基吡啶(0.13g,0.75mmol)和四丁基氯化铵(2mg,0.0375mmol)在5ml50%NaOH水溶液中在100℃加热8小时。在室温冷却该反应混合液,加入5ml水,并用乙酸乙酯萃取。经无水硫酸钠干燥,并浓缩,并通过制备型HPLC,得到8-氯-2-甲基-5-(2-(5-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚。实施例245:2,3,4,5-四氢-2,8-二甲基-5-(2-(1-甲基-1H-吡咯并[2,3-b]吡啶-5-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物427)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、1-甲基-5-乙烯基-1H-吡咯并[2,3-b]吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例246:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(1-甲基-1H-吡咯并[2,3-b]吡啶-5-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物428)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、1-甲基-5-乙烯基-1H-吡咯并[2,3-b]吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例247:2,3,4,5-四氢-2,8-二甲基-5-(2-(5-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物429)将四丁基氯化铵(0.013g,0.0469mmol)溶于2ml50%NaOH水溶液中,在室温搅拌15分钟。然后在其中加入2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.5g,2.49mmol),在室温搅拌10分钟,然后加入3-甲基-5-乙烯基吡啶(0.13g,1.1mmol)。将该反应混合液在120℃搅拌过夜。该反应混合液冷却至室温,用水淬灭,并用乙酸乙酯萃取。经无水硫酸钠干燥有机层,浓缩,并纯化经反相色谱,得到5mg(2,8-二甲基-5-(2-(5-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚),为TFA盐。实施例248:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(5-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物430)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、3-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例249:2,3,4,5-四氢-5-(2-(6-甲氧基吡啶-3-基)乙基)-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物431)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-甲氧基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例250:8-氯-2,3,4,5-四氢-5-(2-(6-甲氧基吡啶-3-基)乙基)-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物432)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-甲氧基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例251:5-(2-(6-乙氧基吡啶-3-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物433)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-甲氧基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例252:8-氯-5-(2-(6-乙氧基吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物434)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、2-甲氧基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例253:5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)-N,N-二甲基吡啶-2-胺的制备(化合物435)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、N,N-二甲基-5-乙烯基吡啶-2-胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例254:5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)-N,N-二甲基吡啶-2-胺的制备(化合物436)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、N,N-二甲基-5-乙烯基吡啶-2-胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例255:N-(5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)-3-甲基吡啶-2-基)乙酰胺的制备(化合物437)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、N-(3-甲基-5-乙烯基吡啶-2-基)乙酰胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例256:N-(5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)-3-甲基吡啶-2-基)乙酰胺的制备(化合物438)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、N-(3-甲基-5-乙烯基吡啶-2-基)乙酰胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例257:5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)-N-异丙基吡啶-2-胺的制备(化合物439)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、N-异丙基-5-乙烯基吡啶-2-胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例258:5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)-N-异丙基吡啶-2-胺的制备(化合物440)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、N-异丙基-5-乙烯基吡啶-2-胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例259:5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)-N-甲基吡啶-2-胺的制备(化合物441)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、N-甲基-5-乙烯基吡啶-2-胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例260:5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)-N-甲基吡啶-2-胺的制备(化合物442)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、N-甲基-5-乙烯基吡啶-2-胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例261:5-(2-(5-氯吡啶-3-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物443)将四丁基氯化铵(0.034g,0.124mmol)溶于15ml50%NaOH水溶液中。在室温搅拌15分钟。然后向其中加入2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.500mg,2.49mmol)。在室温搅拌10分钟。加入3-氯-5-乙烯基吡啶(0.381mg,2.74mmol)。将该反应混合液在100℃加热12-15小时。使该反应混合液在室温冷却,用乙酸乙酯萃取,经无水硫酸钠干燥,浓缩,并经反相色谱纯化,得到纯的化合物的TFA盐(510mg)。将该TFA盐转化为草酸盐(345mg)。实施例262:8-氯-5-(2-(5-氯吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物444)将四丁基氯化铵(0.044g,0.159mmol)溶于10ml50%NaOH水溶液中,并在室温搅拌15分钟。加入8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(700mg,3.18mmol),并在室温进一步搅拌10分钟。加入3-氯-5-乙烯基吡啶(0.486mg,3.49mmol),并将反应混合液在100℃加热12-15小时。在室温冷却该反应混合液,用乙酸乙酯萃取,经无水硫酸钠干燥,浓缩,并经反相色谱纯化,得到TFA盐(750mg)。将TFA盐转化为草酸盐(485mg)。实施例263:5-(2-(5-氟吡啶-3-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物445)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、3-氟-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例264:8-氯-5-(2-(5-氟吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物446)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、3-氟-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例265:N-(5-(2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡啶-2-基)乙酰胺的制备(化合物447)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、N-(5-乙烯基吡啶-2-基)乙酰胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例266:N-(5-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡啶-2-基)乙酰胺的制备(化合物448)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、N-(5-乙烯基吡啶-2-基)乙酰胺和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例267:1-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-2-(6-甲基吡啶-3-基)丙-2-醇的制备(化合物449)将化合物2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(1.5g,7.5mmol,1当量)和NaH(252mg,10.5mmol,1.4当量)在30mlDMF中的混合液加热至120℃达1小时。将其冷却至室温,并历经12分钟滴加在17mlDMF中的化合物2-甲基-5-(2-甲基环氧乙烷-2-基)吡啶(2.46g,16.5mmol,2.2当量)。将温度再次升至120℃,并搅拌3小时。将该反应混合液冷却至室温,并向其中加入5ml水,用700ml乙酸乙酯稀释,并将有机部分用水(100mlX3)洗涤,然后用盐水洗涤。将其经硫酸钠干燥,并在真空下浓缩。经230-400硅胶柱色谱使用10-20%在乙酸乙酯中的甲醇梯度洗脱纯化。收率:2.3g(87%)。实施例268:2,3,4,5-四氢-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)丙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物450)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-(丙-1-烯-2-基)吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例269:2,3,4,5-四氢-2,8-二甲基-5-((1-(6-甲基吡啶-3-基)环丙基)甲基)-1H-吡啶并[4,3-b]吲哚的制备(化合物451)根据通用方法8通过使用5-(1-(溴甲基)环丙基)-2-甲基吡啶、(4-甲基苯基肼盐酸盐)、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例270:2,8-二甲基-5-[2-(6-丙基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物452)向2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.5mmol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.14g,2.5mmol),并将其在室温搅拌10分钟。加入2-丙基-5-乙烯基-吡啶(0.18g,0.5mol),并在100℃再搅拌15小时。反应完成(TLC)后,将反应混合液用水(15mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并减压浓缩。经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)纯化粗品,然后进一步经制备型TLC纯化,得到所需化合物,为黄色油状物(0.015g,收率9%)。将2,8-二甲基-5-[2-(6-丙基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.015g,0.043mmol)溶于THF(1.0mL)中。加入草酸二水合物(0.006g,0.043mmol)在THF(1.0mL)中的溶液,并在室温搅拌30分钟,过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.008g,收率42%)。实施例271:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(吡啶-3-基)乙醇的制备(化合物453)将咔啉(500mg,2.5mmol)溶于DMF(5mL)中。在室温向该溶液中加入NaH(60%,180mg,4.5mmol),并将该反应混合液搅拌10-15分钟,之后加入3-(环氧乙烷-2-基)吡啶(450mg,3.7mmol)。将该反应混合液在室温搅拌4小时(通过LCMS监测)。后处理:将该反应混合液倾至冰水中,并用乙酸乙酯萃取。经硫酸钠干燥有机层,并减压浓缩。经反相HPLC纯化残余物,得到420mg产物,为白色固体(TFA盐)。TLC(硅胶)5:95MeOH:DCM,观察到Rf0.1。实施例272:2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(吡啶-3-基)乙醇的制备(化合物454)将氯代咔啉(Chlorocarboline)(500mg,2.27mmol)溶于DMF中。在室温加入NaH(180mg,4.5mmol),并搅拌10-15分钟。在室温向其中滴加纯的环氧化物(450mg,3.7mmol)。将反应混合物在室温搅拌4小时(通过LCMS监测)。将其倾至冰水中,并用乙酸乙酯萃取,干燥,浓缩,并将残余物经RP-HPLC纯化。得到465mg产物,为白色固体(TFA盐)。TLC5%MeOH-DCM观察到Rf0.1。实施例273:5-(2-(4-氟苯基)丙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物456)在60磅/平方英寸的H2下将化合物5-((Z)-2-(4-氟苯基)丙-1-烯基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚和5-(2-(4-氟苯基)烯丙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(比率1:3,1.8g,5.38mmol,1当量)的混合物与180mgPd/C在50ml甲醇/乙酸(10:1)中的混合液在250mlParrShaker容器中振荡18小时。用甲醇将其经硅藻土垫过滤,并在真空下蒸发。用乙酸乙酯(500ml)稀释残余物,并用50ml饱和的碳酸氢钠溶液洗涤,然后用盐水洗涤。减压浓缩有机部分,并经100-200硅胶柱色谱使用在己烷中的0至50%的乙酸乙酯以5%的间隔洗脱纯化。收率:685mg(38%)。实施例274:2-(4-氟苯基)-1-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)丙-2-醇的制备(化合物457)将化合物2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(2.6g,13.1mmol,1当量)和NaH(55%,750mg,17.2mmol,1.3当量)在60mlTHF中的混合液加热至120℃达1小时。然后将其冷却至室温,并在室温经5分钟滴加在25mlDMF中的化合物2-(4-氟苯基)-2-甲基环氧乙烷(4g,26mmol,2当量),随后120℃加热2小时。将其冷却至室温并加入10ml水,随后用800ml乙酸乙酯稀释,将其先用水(150mlX3)洗涤,然后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩。经230-400硅胶(快速色谱)柱色谱使用在乙酸乙酯中的15%甲醇洗脱纯化。收率:3g(66%)。实施例275:4-(2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)乙酰基)哌嗪-1-甲酸叔丁基酯的制备(化合物458)2-(8-氯-2-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙酸(0.2g,0.719mmol)溶于10mLDCM,加入EDCI.HCl(137mg,0.719mmol),并在室温搅拌10分钟。向该反应混合液中加入N-叔丁氧羰基哌嗪(133mg,0.719mmol),并在室温搅拌24小时。通过TLC和LCMS监测反应。反应完成后,将反应混合液浓缩,并经柱色谱(中性氧化铝;2%甲醇-DCM)纯化,然后经反相色谱纯化,得到90mg4-(2-(8-氯-2-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙酰基)哌嗪-1-甲酸叔丁基酯。实施例276:2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-1-(4-甲基哌嗪-1-基)乙酮的制备(化合物459)将2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙酸(0.57g,2.2mmol)溶于DCM中,冷却至0℃,加入草酰氯(0.228mL,2.6mmol),随后加入DMAP(催化量),并在室温搅拌1小时。将溶剂在氮气下蒸发,在室温缓慢滴加在DCM中的N-甲基哌嗪(0.269mg,2.4mmol)和DMAP(80mg)(在氮气下),并在室温搅拌30分钟。将该反应混合液用水淬灭,用10%NaHCO3溶液中和,并用乙酸乙酯萃取。经无水硫酸钠干燥有机层,并蒸发至干燥。将粗品产物经反相色谱纯化,得到100mg2-(2,8-二甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)-1-(4-甲基哌嗪-1-基)乙酮,为TFA盐。实施例277:8-氯-5-(2-(4-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物460)由8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、4-(三氟甲基)-3-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例278:5-(2-(4-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物461)由2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚、4-(三氟甲基)-3-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例279:8-乙基-5-(2-(4-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物462)由8-乙基-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚、4-(三氟甲基)-3-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例280:5-(2-(4-(三氟甲基)吡啶-3-基)乙基)-2,3,4,5-四氢-8-异丙基-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物463)由2,3,4,5-四氢-8-异丙基-2-甲基-1H-吡啶并[4,3-b]吲哚、4-(三氟甲基)-3-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例281:8-氯-5-(2-(2-乙基-1H-吡咯-1-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚的制备(化合物464)根据方法8通过使用1-(2-溴乙基)-2-乙基-1H-吡咯、(4-氯苯基肼盐酸盐)、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例282:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(2,4-二甲基-1H-吡咯-1-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物465)根据方法8通过使用1-(2-溴乙基)-2,4-二甲基-1H-吡咯、(4-氯苯基肼盐酸盐)、三乙胺和N-甲基-4-哌啶酮制备标题化合物。实施例283:5-(2-(1,2,3,4-四氢-8-(羟基甲基)-2-甲基吡啶并[4,3-b]吲哚-5-基)乙基)吡啶-2-甲酸的制备(化合物466)由(2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚-8-基)甲醇、5-乙烯基吡啶-2-甲酸和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例284:2,3,4,5-四氢-5-(2-(6-(羟基甲基)吡啶-3-基)乙基)-2-甲基-1H-吡啶并[4,3-b]吲哚-8-甲酸的制备(化合物467)由2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚-8-甲酸、(5-乙烯基吡啶-2-基)甲醇和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例285:(5-(2-(8-(羟基甲基)-2-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)乙基)吡啶-2-基)甲醇的制备(化合物468)由(2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚-8-基)甲醇、(5-乙烯基吡啶-2-基)甲醇和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例286:5-(2-(6-羧基吡啶-3-基)乙基)-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚-8-甲酸的制备(化合物469)由2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚-8-甲酸、5-乙烯基吡啶-2-甲酸和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例287:8-氯-2,3,4,5-四氢-2-甲基-5-(2-(6-丙基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物476)向8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.1g,0.00045mol)在N-甲基2-吡咯烷酮(1.0mL)中的溶液中加入粉末状的氢氧化钾(0.20g,0.0036mol),并将其在室温搅拌10分钟。加入2-丙基-5-乙烯基-吡啶(0.2g,0.00136mol),并在100℃再搅拌24小时。反应完成(TLC)后,将反应混合液用水(10mL)稀释,并用乙酸乙酯(3x50mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。将粗品经柱色谱(7%MeOH:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)半-纯化,然后进一步经制备型TLC纯化,得到所需化合物,为黄色油状物(0.016g,收率9.6%)。将8-氯-5-[2-(6-异丙基-吡啶-3-基)-乙基]-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.016g,0.0000435mol)溶于THF(1.0mL)中。加入草酸二水合物(0.005g,0.00007mol)在THF(0.5mL)中的溶液,并在室温搅拌30分钟。过滤得到的沉淀物,并干燥,得到草酸盐,为灰白色固体(0.010g,收率52%)。实施例288:7-氮杂-2-甲基-5-(2-(吡啶-2-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物481)向四正-丁基氯化铵(0.0075g,0.04mmol)在50%NaOH水溶液(3mL)中的溶液中加入7-氮杂-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.075g,0.4mmol)和2-乙烯基吡啶(0.047g,0.44mmol)。然后在90℃加热7小时。通过LCMS、TLC监测该反应。反应完成后,将混合物用水淬灭,并用乙酸乙酯萃取。经硫酸钠干燥合并的有机层,并减压浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到4mg产物,为TFA盐。实施例289:9-氮杂-2-甲基-5-(2-(吡啶-4-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物482)向四正-丁基氯化铵(0.017g,0.09mmol)在50%NaOH水溶液(5mL)中的溶液中加入9-氮杂-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.17g,0.9mmol)和4-乙烯基吡啶(0.107g,1mmol)。然后在90℃加热7小时。通过LCMS、TLC监测该反应。反应完成后,将混合物用水淬灭,并用乙酸乙酯萃取。经硫酸钠干燥合并的有机层,并减压浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到3mg产物,为TFA盐。实施例290:7-氮杂-2-甲基-5-(2-(吡啶-4-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物483)向四正-丁基氯化铵(0.017g,0.09mmol)在50%NaOH水溶液(5mL)中的溶液中加入7-氮杂-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.17g,0.9mmol)和4-乙烯基吡啶(0.107g,1mmol),然后在90℃加热7小时。通过LCMS、TLC监测该反应。反应完成后,将混合物用水淬灭,并用乙酸乙酯萃取。经硫酸钠干燥合并的有机层,并减压浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到5mg产物,为TFA盐。实施例291:9-氮杂-2-甲基-5-(2-(吡啶-2-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物484)向四正-丁基氯化铵(0.0075g,0.04mmol)在50%NaOH水溶液(3mL)中的溶液中加入9-氮杂-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.075g,0.4mmol)和2-乙烯基吡啶(0.047g,0.44mmol),然后在90℃加热7小时。通过LCMS、TLC监测该反应。反应完成后,将混合物用水淬灭,并用乙酸乙酯萃取。经硫酸钠干燥合并的有机层,并减压浓缩,经反相色谱(C-18,500mmx50mm,流动相A=0.05%TFA的水溶液,B=0.05%TFA的乙腈溶液,梯度:在30分钟内,10%B至80%B,进样体积:5mL)纯化后,得到2mg产物,为TFA盐。实施例292:2,3,4,5-四氢-8-碘-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-1H-吡啶并[4,3-b]吲哚的制备(化合物485)向8-碘-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.5g.0.0016mol)在N-甲基2-吡咯烷酮(5mL)中的溶液中加入粉末状的氢氧化钾(0.9g,0.016mol),并将其在室温搅拌10分钟。加入2-甲基-5-乙烯基-吡啶(0.969g,0.0048mol),并在100℃再搅拌24小时,反应完成后(TLC),将反应混合液用水(50mL)稀释,并用乙酸乙酯(3x150mL)萃取。经无水硫酸钠干燥有机层,并使用旋转蒸发器减压浓缩。将粗品经柱色谱(6%甲醇:DCM,硅胶100-200目,柱直径-2.5cm,硅胶高度-大约5英寸)半-纯化,然后进一步经制备型HPLC纯化,得到所需化合物,为褐色固体(0.105g,收率15.2%)。将8-碘-2-甲基-5-[2-(6-甲基-吡啶-3-基)-乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.095g,0.00022mol)溶于THF(2.0mL)中。加入草酸二水合物(0.0277g,0.00022mol)在THF(2.0mL)中的溶液,并在室温搅拌30分钟。过滤沉淀物,并干燥,得到草酸盐,为灰白色固体(0.09g,收率79%)。实施例293:2-(1,2,3,4-四氢-8-碘-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(哌啶-1-基)乙酮的制备(化合物486)用己烷洗涤氢化钠以除去油,并在真空下干燥。然后将氢化钠溶于THF中。在0℃向该溶液中滴加在THF中的8-碘-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(100mg,0.32mmol)。然后将该反应混合液搅拌0.5小时。向反应混合液中滴加2-氯-1-(哌啶-1-基)乙酮在THF中的溶液。然后在室温搅拌该反应混合物2小时。通过TLC监测反应,反应完成后,用冰水淬灭反应混合液。将THF蒸发并用乙酸乙酯萃取水层。经无水硫酸钠干燥有机层。经柱色谱纯化粗品化合物,得到25mg所需化合物。将其中的5mg与乙醇HCl搅拌,得到2-(8-碘-2-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)-1-(哌啶-1-基)乙酮的HCl盐。实施例294:8-溴-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物487)将四丁基氯化铵(0.026g,0.094mmol)溶于15ml50%NaOH水溶液(7ml)中,并在室温搅拌15分钟。加入8-溴-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(0.500mg,1.89mmol),并在室温进一步搅拌10分钟。加入2-甲基-5-乙烯基吡啶(0.247mg,2.08mmol),并将反应混合液在100℃加热12-15小时。在室温冷却该反应混合液,用乙酸乙酯萃取,经无水硫酸钠干燥,浓缩,并经反相HPLC纯化,得到63mg8-溴-2-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚,为TFA盐。实施例295:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(4-甲氧基苯基)丙-2-醇的制备(化合物488)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(4-甲氧基苯基)-2-甲基环氧乙烷(400mg,2.43mmol,1.85当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备分析样本。实施例296:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(6-(三氟甲基)吡啶-3-基)丙-2-醇的制备(化合物489)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(三氟甲基)-5-(2-甲基环氧乙烷-2-基)吡啶(400mg,1.97mmol,1.5当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例297:1-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-2-(吡啶-3-基)丙-2-醇的制备(化合物490)向2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.4mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.14当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加3-(2-甲基环氧乙烷-2-基)吡啶(400mg,2.96mmol,2.1当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层并用乙酸乙酯(1X20ml)萃取水层。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例298:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(吡啶-3-基)丙-2-醇的制备(化合物491)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.3mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加3-(2-甲基环氧乙烷-2-基)吡啶(400mg,2.96mmol,2.3当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例299:1-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)-2-(吡啶-4-基)丙-2-醇的制备(化合物492)向2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.4mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.14当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加4-(2-甲基环氧乙烷-2-基)吡啶(400mg,2.96mmol,2.1当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例300:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(吡啶-4-基)丙-2-醇的制备(化合物493)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.3mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加4-(2-甲基环氧乙烷-2-基)吡啶(400mg,2.96mmol,2.3当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层并用乙酸乙酯(1X20ml)萃取水层。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例301:1-(5-(三氟甲基)吡啶-3-基)-2-(1,2,3,4-四氢-2,8-二甲基吡啶并[4,3-b]吲哚-5-基)乙醇的制备(化合物494)向2,3,4,5-四氢-2,8-二甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.44mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.1当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加3-(三氟甲基)-5-(环氧乙烷-2-基)吡啶(400mg,2.11mmol,1.5当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例302:2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(5-(三氟甲基)吡啶-3-基)乙醇的制备(化合物495)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加3-(三氟甲基)-5-(环氧乙烷-2-基)吡啶(400mg,2.1mmol,1.6当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例303:2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-(3-氟-4-甲氧基苯基)乙醇的制备(化合物496)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(3-氟-4-甲氧基苯基)环氧乙烷(400mg,2.37mmol,1.8当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例304:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(6-丙基吡啶-3-基)丙-2-醇的制备(化合物497)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加5-(2-甲基环氧乙烷-2-基)-2-丙基吡啶(400mg,2.26mmol,1.72当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例305:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(3,4-二氟苯基)丙-2-醇的制备(化合物498)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(3,4-二氟苯基)-2-甲基环氧乙烷(400mg,2.4mmol,1.8当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例306:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(4-氯苯基)丙-2-醇的制备(化合物499)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(4-氯苯基)-2-甲基环氧乙烷(400mg,2.4mmol,1.8当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例307:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(4-氯-3-氟苯基)丙-2-醇的制备(化合物500)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(4-氯-3-氟苯基)-2-甲基环氧乙烷(400mg,2.15mmol,1.6当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例308:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(3,4-二氯苯基)丙-2-醇的制备(化合物501)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(3,4-二氯苯基)-2-甲基环氧乙烷(400mg,1.97mmol,1.5当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例309:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(3-氯-4-氟苯基)丙-2-醇的制备(化合物502)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(3-氯-4-氟苯基)-2-甲基环氧乙烷(400mg,2.14mmol,1.6当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例310:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(2,4-二氟苯基)丙-2-醇的制备(化合物503)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(2,4-二氟苯基)-2-甲基环氧乙烷(400mg,2.4mmol,1.8当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例311:1-(8-氟-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(4-氟苯基)丙-2-醇的制备(化合物504)向8-氟-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(4-氟苯基)-2-甲基环氧乙烷(400mg,2.63mmol,2.0当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例312:1-(8-氯-2-环丙基-1,2,3,4-四氢吡啶并[4,3-b]吲哚-5-基)-2-(4-氟苯基)丙-2-醇的制备(化合物505)向8-氯-2-环丙基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(290mg,1.18mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.36当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(4-氟苯基)-2-甲基环氧乙烷(400mg,2.63mmol,2.2当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例313:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-苯基丙-2-醇的制备(化合物506)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-甲基-2-苯基环氧乙烷(400mg,2.98mmol,2.3当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层并用乙酸乙酯(1X20ml)萃取水层。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例314:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(2,4,6-三氟苯基)丙-2-醇的制备(化合物507)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(2,4,6-三氟苯基)-2-甲基环氧乙烷(400mg,2.1mmol,1.6当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例315:3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(4-氟苯基)丁-2-醇的制备(化合物508)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(4-氟苯基)-2,3-二甲基环氧乙烷(400mg,2.4mmol,1.8当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例316:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(2,4-二氯苯基)丙-2-醇的制备(化合物509)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(2,4-二氯苯基)-2-甲基环氧乙烷(400mg,1.96mmol,1.5当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例317:1-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-2-(4-氟苯基)丁-2-醇的制备(化合物510)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-乙基-2-(4-氟苯基)环氧乙烷(400mg,2.4mmol,1.8当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层并用乙酸乙酯(1X20ml)萃取水层。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例318:3-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1,1,1-三氟-2-(4-氟苯基)丙-2-醇的制备(化合物511)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-(三氟甲基)-2-(4-氟苯基)环氧乙烷(400mg,1.94mmol,1.48当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例319:2-(8-氯-1,2,3,4-四氢-2-甲基吡啶并[4,3-b]吲哚-5-基)-1-环丙基-1-(4-氟苯基)乙醇的制备(化合物512)向8-氯-2,3,4,5-四氢-2-甲基-1H-吡啶并[4,3-b]吲哚(290mg,1.31mmol,1.0当量)在DMF(6ml)中的溶液中加入氢化钠(38mg,1.6mmol,1.2当量),并搅拌着加热至120℃达1小时。将该反应混合液冷却至0℃,并历经5分钟滴加2-环丙基-2-(4-氟苯基)环氧乙烷(400mg,2.2mmol,1.71当量)。将温度升至120℃,并搅拌2小时。将该反应混合液冷却至室温,并在乙酸乙酯(60ml)和水(15ml)之间分配。分离有机层,并将水层用乙酸乙酯(1X20ml)萃取。将合并的有机层用水洗涤,随后用盐水洗涤,经硫酸钠干燥,并在真空下浓缩,得到粗品产物。将产物通过快速柱色谱经硅胶(230-400目,用1%三乙胺/己烷去活化)使用5至15%甲醇/乙酸乙酯梯度洗脱纯化,得到游离碱。将纯品化合物转化为其草酸盐。通过将游离碱溶于10mLTHF中,并用1当量的草酸二水合物处理来制备所述分析样品。实施例320:2-(8-溴-2-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)-1-(哌啶-1-基)乙酮的制备(化合物516)用己烷洗涤氢化钠(0.6g,15mmol)以除去油,并在真空下干燥。然后将氢化钠溶于THF中。在0℃向该溶液中滴加在THF中的8-溴-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(2g,7.5mmol)。然后将反应混合液搅拌0.5小时。向反应混合液中滴加2-氯-1-(哌啶-1-基)乙酮(1.8g,11.3mmol)在THF中的溶液。然后将反应混合液在室温搅拌2小时。通过TLC监测反应。反应完成后,用冰水淬灭反应混合液。将THF蒸发,并用乙酸乙酯萃取水层。经无水硫酸钠干燥有机层。用己烷和乙醚洗涤粗品化合物以除去有颜色的杂质,然后通过使用甲醇重结晶,得到1.5g所需化合物,然后将其中的0.8g化合物在乙醇HCl中搅拌,得到2-(8-溴-2-甲基-3,4-二氢-1H-吡啶并[4,3-b]吲哚-5(2H)-基)-1-(哌啶-1-基)乙酮的HCl盐。实施例321:8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-3,4-二氢-1H-吡啶并[4,3-b]吲哚-2(5H)-甲醛的制备(化合物517)由3,4-二氢-8-甲基-1H-吡啶并[4,3-b]吲哚-2(5H)-甲醛、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例322:2-丁基-8-甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚的制备(化合物518)由2-丁基-2,3,4,5-四氢-8-甲基-1H-吡啶并[4,3-b]吲哚、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例323:3,4-二氢-3-羟基-2,8-二甲基-5-(2-(6-甲基吡啶-3-基)乙基)-2H-吡啶并[4,3-b]吲哚-1(5H)-酮的制备(化合物316)由3,4-二氢-3-羟基-2,8-二甲基-2H-吡啶并[4,3-b]吲哚-1(5H)-酮、2-甲基-5-乙烯基吡啶和KOH(5-7当量)在NMP中的混合液在25℃至100℃的温度范围内制备标题化合物。经制备型HPLC分离得到产物。实施例324.1,2,4-二唑G6-a-f的制备使用流程I中所述的合成路线制备1,2,4-二唑类化合物(G6)。根据标准方法将通过Fischer反应制备的吡啶并[4,3-b]吲哚类化合物(3)烷基化,得到(4)。将腈类化合物(4)转化为其相应的羟基脒类化合物(5)。在NaOEt的存在下用甲酸乙酯环化5,得到G6。流程I由相应的腈类化合物(G4)制备羟基脒类化合物(G5):将溶于水中的氢氧化钠(1当量)加入溶于95%乙醇中的盐酸羟胺(1当量)溶液中。将在95%乙醇中的适合的腈(0.5当量)加入以上溶液中。将该混合液在回流下加热6-7小时,冷却,并用稀释水。通过过滤分离固体,用水、乙醇和乙醚洗涤,并由MeOH重结晶。由相应的羟基脒类化合物(G5)制备1,2,4-二唑类化合物(G6):将适合的羟基脒(0.6-1当量)、苯甲酸乙酯(1当量)和乙醇钠(1当量)的溶液在无水乙醇中在回流下加热7-15小时。减压除去溶剂,并将残余物在CHCl3和水之间分配。用水洗涤有机层,干燥(Na2SO4)和蒸发。将残余物经中性氧化铝色谱(EtOAc/Et3N,8:1)纯化。3-(2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙腈(G4-a):向吡啶并[4,3-b]吲哚G3-a(R19=H)(2.108g,11.3mmol)在苯(30mL)和丙烯腈(10mL)的混合液中的溶液中加入苄基三甲基氢氧化铵溶液(在甲醇中40重量%,0.7mL)。将该混合液在回流下加热1小时,冷却,用水洗涤,经Na2SO4干燥,并浓缩。经硅胶柱色谱(苯/Et3N,9:1)纯化,得到2.174g(80%)标题化合物,mp108-110℃(丙酮,-12℃)。1HNMR(丙酮-d6):2.46(3H,s),2.73-3.01(6H,m),3.56(2H,s),4.48(2H,t),6.97-7.16(2H,m),7.38(1H,d),7.45(1H,d)。由吡啶并[4,3-b]吲哚G3-b(R19=Cl)制备了3-(8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙腈(G4-b),收率76%,mp127-129℃(丙酮,-12℃)。1HNMR(丙酮-d6):2.50(3H,s),2.75-3.06(6H,m),3.58(2H,s),4.54(2H,t),7.12(1H,dd),7.43(1H,d),7.53(1H,d)。以72%收率制备了N-羟基-3-(2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙脒(G5-a),mp218-219.5℃(分解)。1HNMR(DMSO-d6/CDCl3,2:1):2.34(2H,t),2.48(3H,s),2.70-2.92(4H,m),3.52(2H,s),4.22(2H,t),5.48(2H,s),6.90-7.10(2H,m),7.28(1H,d),7.40(1H,d),8.90(1H,s)。以65%收率得到N-羟基-3-(8-甲氧基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙脒(G5-b),mp211-213℃(分解)。1HNMR(DMSO-d6/CDCl3,2:1):2.36(2H,t),2.48(3H,s),2.68-2.84(4H,m),3.52(2H,s),3.79(3H,s),4.21(2H,t),5.39(2H,s),6.67(1H,dd),6.77(1H,d),7.27(1H,d),8.90(1H,s)。以75%收率制备了3-(2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)-N-羟基丙脒(G5-c),mp218-220℃(分解)。1HNMR(DMSO-d6/CDCl3,2:1):2.36(5H,m),2.46(3H,s),2.68-2.88(4H,m),3.49(2H,s),4.22(2H,t),5.48(2H,s),6.90(1H,d),7.10(1H,s),7.30(1H,d),8.92(1H,s)。以68%收率制备了3-(8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)-N-羟基丙脒(G5-d),mp222-224℃(分解)。1HNMR(DMSO-d6/CCl4,3:1):2.32(2H,t),2.47(3H,s),2.73-2.92(4H,m),3.54(2H,s),4.23(2H,t),5.50(2H,s),7.03(1H,dd),7.32(1H,d),7.46(1H,d),8.87(1H,s)。以71%收率制备了3-(8-氟-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)-N-羟基丙脒(G5-f),mp222-224℃(分解)。1HNMR(DMSO-d6/CCl4,3:1):2.35(2H,t),2.45(3H,s),2.72-2.92(4H,m),3.53(2H,s),4.25(2H,t),5.55(2H,s),6.89(1H,td),7.08(1H,dd),7.44(1H,dd),8.89(1H,s)。以1.0-当量的规模制备了2-甲基-5-[2-(5-苯基-1,2,4-二唑-3-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G6-a)。经色谱纯化,得到60%(G6-a),mp111-112℃(丙酮/戊烷)。1HNMR(DMSO-d6/CDCl3,1:2):2.48(3H,s),2.70-2.88(4H,m),3.15(2H,t),3.55(2H,s),4.48(2H,t),6.90-7.08(2H,m),7.30(2H,d),7.48-7.65(3H,m),8.80(2H,d)。盐酸盐(G6-a),mp软化218℃,226-228℃(分解)(H2O)。以1.0-当量的规模制备了8-甲氧基-2-甲基-5-[2-(5-苯基-1,2,4-二唑-3-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G6-b)。经色谱纯化,得到50.6%(G6-b),mp77.5-79℃(Et3N,-12℃)。1HNMR(CDCl3):2.60(3H,s),2.82-2.95(4H,m),3.22(2H,t),3.68(2H,s),3.88(3H,s),4.50(2H,t),6.78-6.94(2H,m),7.28(1H,d),7.52-7.70(3H,m),8.18(2H,d)。盐酸盐(G6-b),mp137-138℃(H2O)。以1.0-当量的规模制备了2,8-二甲基-5-[2-(5-苯基-1,2,4-二唑-3-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]-吲哚(G6-c)。经色谱纯化,得到48%(G6-c),mp120-120.5℃(EtOAc/戊烷)。1HNMR(DMSO-d6/CDCl3,1:2):2.40(3H,s),2.48(3H,s),2.72-2.90(4H,m),3.17(2H,t),3.57(2H,s),4.50(2H,t),6.92(1H,d),7.13(1H,s),7.22(1H,d),7.52-7.71(3H,m),8.11(2H,d)。盐酸盐(G6-c),mp软化213℃,220-223℃(分解)(H2O)。以0.6-当量的规模制备了8-氯-2-甲基-5-[2-(5-苯基-1,2,4-二唑-3-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G6-d)。经色谱纯化,得到64%(G6-d),mp122-123℃(EtOAc/戊烷)。1HNMR(CDCl3):2.58(3H,s),2.83-2.98(4H,m),3.23(2H,t),3.66(2H,s),4.52(2H,t),7.13(1H,dd),7.31(1H,m),7.42(1H,d),7.53-7.70(3H,m),8.17(2H,d)。盐酸盐(G6-d),mp软化214℃,227-229℃(分解)(H2O)。2-甲基-5-[2-(5-吡啶-4-基-1,2,4-二唑-3-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G6-e)。向乙醇钠(得自0.188gNa和25mL无水乙醇)中加入异烟酸乙酯(1.249g,8.3mmol)和5-a(1.336g,4.9mmol)。将该混合液在回流下加热7小时。减压除去溶剂,并将残余物在乙酸乙酯和水之间分配。用水洗涤有机层,经Na2SO4干燥和蒸发。将残余物经硅胶色谱(苯/Et3N,9:1)纯化,得到1.145g(64.9%)的G6-e,mp110.5-111.5℃(i-PrOH,-12℃)。1HNMR(DMSO-d6/CDCl3,2:1):2.42(3H,s),2.64-2.90(4H,m),3.25(2H,t),3.51(2H,s),4.53(2H,t),6.90-7.12(2H,m),7.26-7.42(2H,m),7.98(2H,d),8.87(2H,d)。以0.6-当量的规模制备了8-氟-2-甲基-5-[2-(5-苯基-1,2,4-二唑-3-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G6-f)。经色谱纯化,得到76%(G6-f),mp121.5-122.5℃(EtOAc/戊烷)。1HNMR(DMSO-d6/CDCl3,1:2):2.47(3H,s),2.73-2.92(4H,m),3.20(2H,t),3.55(2H,s),4.51(2H,t),6.82(1H,td),7.00(1H,dd),7.29(1H,dd),7.50-7.71(3H,m),8.11(2H,d)。盐酸盐(G6-f),mp187℃,222-224℃(分解)(H2O)。实施例325.噻唑类化合物G8-a-f的制备使用流程II中所示的合成路线制备噻唑类化合物(G8)。将腈类化合物(G4)转化为其相应的硫代丙酰胺类化合物(7)。用2-溴酮(R14a-COCH2Br)环化7,得到G8。流程II3-(2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)硫代丙酰胺(G7-a)。将G4-a(3.0g,12.5mmol)在吡啶(25mL)和三乙胺(3.5mL)中的溶液用H2S饱和15分钟。在室温72小时后在真空下浓缩该反应混合液。用NaOH水溶液以及用水洗涤固体残余物。在真空下干燥后,得到3.3g(96%)的G7-a,mp149-151℃。1HNMR(DMSO-d6/CDCl3,1:2):2.53(3H,s),2.75-3.01(6H,m),3.60(2H,s),4.46(2H,t),6.94-7.16(2H,m),7.34(1H,d),7.43(1H,d),9.12(2H,s)。通过以上的对G7-a的方法制备其它的硫代丙酰胺类化合物7。以89%收率制备了3-(8-甲氧基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)硫代丙酰胺(G7-b),mp145-147℃。1HNMR(DMSO-d6/CDCl3,1:2):2.51(3H,s),2.74-2.98(6H,m),3.56(2H,s),3.80(3H,s),4.41(2H,t),6.72(1H,d),6.80(1H,s),7.31(1H,d),9.09(2H,s)。以91%收率制备了3-(2,8-二甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)硫代丙酰胺(7G-c),mp161-164℃。1HNMR(DMSO-d6/CDCl3,1:2):2.40(3H,s),2.52(3H,s),2.75-2.99(6H,m),3.57(2H,s),4.42(2H,t),6.92(1H,d),7.13(1H,s),7.31(1H,d),9.10(2H,s)。以91%收率制备了3-(8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)硫代丙酰胺(G7-e),mp156-158℃。1HNMR(DMSO-d6):2.41(3H,s),2.66-2.94(6H,m),3.47(2H,s),4.41(2H,t),7.06(1H,d),7.38(1H,s),7.46(1H,d),9.30(1H,s),9.51(1H,s)。以94%收率制备了3-(8-氟-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)硫代丙酰胺(G7-f),mp165-167℃。1HNMR(DMSO-d6/CDCl3,1:2):2.52(3H,s),2.75-3.00(6H,m),3.55(2H,s),4.44(2H,t),6.84(1H,td),6.99(1H,dd),7.38(1H,dd),9.13(2H,s)。2-甲基-5-[2-(4-苯基噻唑-2-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G8-a)。将G7-a(1.62g,5.9mmol)、2-溴苯乙酮(1.18g,5.9mmol)和在乙醇(22mL)中的5%HCl的混合液在回流下加热20分钟。将该反应混合液冷却至室温,并在真空下浓缩。将残余物在CH2Cl2和NaOH水溶液之间分配。用水洗涤有机层,干燥(Na2SO4),并蒸发。经硅胶柱色谱纯化(苯/Et3N,13:1),得到1.79g(80.9%)标题化合物,mp142-143(i-PrOH)。1HNMR(DMSO-d6/CDCl3,1:2):2.48(3H,s),2.73(4H,s),3.43(2H,t),3.58(2H,s),4.53(2H,t),6.95-7.15(2H,m),7.27-7.53(6H,m),7.90(2H,d)。盐酸盐(G8-a),mp128-130℃(H2O)。通过以上的对G8-a的方法制备其它的被取代的噻唑类化合物8。以62.8%收率制备了8-甲氧基-2-甲基-5-[2-(4-苯基噻唑-2-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G8-b),mp135-136.5℃(庚烷)。HNMR(丙酮-d6):2.43(3H,s),2.61-2.77(4H,m),3.45-3.57(4H,m),3.82(3H,s),4.58(2H,t),6.75(1H,dd),6.93(1H,d),7.30-7.53(4H,m),7.74(1H,s),8.04(2H,d)。盐酸盐(G8-b),mp162-163℃(H2O)。以72%收率制备了2,8-二甲基-5-[2-(4-苯基噻唑-2-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G8-c),mp148.5-149℃(EtOAc)。1HNMR(DMSO-d6/CDCl3,1:2):2.41(3H,s),2.45(3H,s),2.69(4H,s),3.39(2H,t),3.53(2H,s),4.49(2H,t),6.87(1H,d),7.06-7.20(2H,m),7.24-7.45(4H,m),7.88(2H,d)。盐酸盐(G8-c),mp127-129℃(H2O)。2-甲基-5-[2-(4-吡啶-4-基-噻唑-2-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G8-d)。将7-a(1.0g,3.7mmol)、2-溴-1-吡啶-4-基-乙酮氢溴酸盐(1.029g,3.7mmol)和在乙醇(44mL)中的5%HCl的混合液在回流下加热20分钟。将该反应混合液冷却至室温,并在真空下浓缩。将残余物在CH2Cl2和NaOH水溶液之间分配。用水洗涤有机层,经Na2SO4干燥和蒸发。用丙酮洗涤固体残余物。从2-丙醇中重结晶,得到G8-d(0.42g,30.6%),mp158.5-159.5℃。1HNMR(DMSO-d6/CCl4,1:1):2.42(3H,s),2.67(4H,s),3.44(2H,t),3.53(2H,s),4.52(2H,t),6.90-7.09(2H,m),7.27-7.40(2H,m),7.86(2H,d),8.17(1H,s),8.58(2H,d)。8-氯-2-甲基-5-[2-(4-苯基噻唑-2-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G8-e)。将7-e(1.033g,3.4mmol)、2-溴苯乙酮(0.67g,3.4mmol)和在乙醇(20mL)中的5%HCl的混合液在回流下加热20分钟。将该反应混合液冷却至室温,并在真空下浓缩。将残余物在CH2Cl2和NaOH水溶液之间分配。用水洗涤有机层,经Na2SO4干燥,并蒸发。经硅胶柱色谱(苯/Et3N,9:1)纯化,得到1.019g(74%)的G8-e,mp140-142℃(i-PrOH)。1HNMR(丙酮-d6):2.42(3H,s),2.56-2.81(4H,m),3.39-3.60(4H,m),4.60(2H,t),7.04(1H,d),7.27-7.51(5H,m),7.71(1H,s),7.98(2H,d)。以77%收率制备了8-氟-2-甲基-5-[2-(4-苯基噻唑-2-基)乙基]-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚(G8-f),mp136.5-137℃(EtOAc)。1HNMR(DMSO-d6/CDCl3,1:2):2.49(3H,s),2.74(4H,s),3.42(2H,t),3.55(2H,s),4.53(2H,t),6.82(1H,td),7.01(1H,dd),7.20-7.53(5H,m),7.90(2H,d)。盐酸盐(G8-f),mp135-137℃(H2O)。实施例326.5-被取代的-2,4-二氢-1,2,4-三唑-3-硫酮类化合物G11-a-d的制备使用流程III所示的合成路线制备5-被取代的-2,4-二氢-1,2,4-三唑-3-硫酮类化合物(G11)。将吡啶并[4,3-b]吲哚类化合物(3)烷基化,得到酯类化合物(G9),将其转化为其相应的酰肼类化合物(G10)。羧酸酰肼类化合物(G10)和异硫氰酸甲酯反应,得到2,4-二氢-1,2,4-三唑-3-硫酮类化合物(G11)。流程III3-(2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙酸乙酯(G9-a)。将苄基三甲基氢氧化铵溶液(在甲醇中40重量%,3.5mL)加入G3-a(17.114g,92mmol)在苯(170mL)和丙烯酸乙酯(75mL)中的混合液中的溶液。将该混合液在回流下加热7小时,冷却,用水洗涤,并用2MHCl萃取。用苯萃取该酸性的溶液,用10%NaOH碱化,并用苯萃取。用水洗涤合并的萃取物,干燥,并蒸发为油状物(21.18g,80%)。1HNMR(丙酮-d6):1.13(3H,t),2.49(3H,s),2.70-2.96(6H,m),3.60(2H,s),4.05(2H,q),4.37(2H,t),6.94-7.15(2H,m),7.32-7.43(2H,m)。通过以上的对9-a的方法制备其它的被取代的酯类化合物9。以88%收率制备了3-(8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙酸乙酯(G9-b),油状物。1HNMR(丙酮-d6):1.13(3H,t),2.45(3H,s),2.69-2.98(6H,m),3.52(2H,s),4.03(2H,q),4.39(2H,t),7.06(1H,dd),7.36(1H,d),7.40(1H,d)。以89%收率制备了3-(8-氟-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙酸乙酯(G9-c),油状物。1HNMR(丙酮-d6):1.13(3H,t),2.45(3H,s),2.68-2.94(6H,m),3.51(2H,s),4.03(2H,q),4.38(2H,t),6.86(1H,td),7.06(1H,dd),7.37(1H,dd)。以80%收率制备了3-(8-甲氧基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙酸乙酯(G9-d),油状物。1HNMR(丙酮-d6):1.15(3H,t),2.46(3H,s),2.65-2.94(6H,m),3.51(2H,s),3.78(3H,s),4.05(2H,q),4.34(2H,t),6.73(1H,d),6.89(1H,s),7.28(1H,d)。3-(2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙酰肼(G10-a)。向G9-a(20.356g,74.8mmol)在乙醇(50mL)中的溶液中加入肼一水合物(20mL,0.41mol),并将混合液在回流下加热2小时30分钟。在-12℃放置72小时后,收集得到的结晶,用2-丙醇洗涤,并干燥,得到12.7g(65.6%)G10-a,mp123-124℃.1HNMR(DMSO-d6/CCl4,3:1):2.32-2.47(5H,m),2.63-2.92(4H,m),3.50(2H,s),4.11(2H,brs),4.26(2H,t),6.89-7.12(2H,m),7.24-7.42(2H,m),9.03(1H,brs)。通过以上的对G10-a的方法制备其它的酰肼类化合物G10。以74%收率制备了3-(8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙酰肼(G10-b),mp172-173℃。1HNMR(DMSO-d6/CDCl3,1:1):2.38-2.50(5H,m),2.68-2.94(4H,m),3.50(2H,s),4.00(2H,brs),4.28(2H,t),7.01(1H,dd),7.28(1H,d),7.32(1H,d),9.05(1H,brs)。以72%收率制备了3-(8-氟-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙酰肼(G10-c),mp158-159℃。1HNMR(DMSO-d6/CCl4,2:1):2.30-2.45(5H,m),2.67-2.92(4H,m),3.48(2H,s),3.91(2H,brs),4.25(2H,t),6.82(1H,td),7.00(1H,dd),7.32(1H,dd),9.01(1H,brs)。3-(8-甲氧基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)丙酰肼(G10-d)。将9-d(20.872g,66mmol)在乙醇(50mL)和肼一水合物(20mL,0.41mol)中的溶液在回流下加热2小时15分钟,然后在真空下蒸发。残余物在乙醇(-12℃)中结晶,得到8.9g(44.6%)G10-d,mp126.5-127.5℃.1HNMR(DMSO-d6/CDCl3,1:1):2.36-2.49(5H,m),2.67-2.94(4H,m),3.52(2H,s),3.77(3H,s),4.00(2H,brs),4.26(2H,t),6.69(1H,d),6.78(1H,s),7.22(1H,d),9.04(1H,brs)。4-甲基-5-[2-(2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)乙基]-2,4-二氢-1,2,4-三唑-3-硫酮(G11-a)。将G10-a(2.187g,8mmol)、异硫氰酸甲酯(0.587g,8mmol)和55mL乙醇的混合液在回流下加热6小时30分钟,然后在室温放置48小时。通过过滤收集得到的结晶,用乙醇洗涤,并干燥,得到2.268g(86%)G11-a,mp216-219℃(EtOH)。1HNMR(DMSO-d6/CDCl3,1:1):2.45(3H,s),2.72(4H,s),3.05(2H,t),3.16(3H,s),3.53(2H,s),4.42(2H,t),6.88-7.13(2H,m),7.22-7.38(2H,m),13.52(1H,brs);MS,m/z327(M+)。通过以上的对G11-a的方法制备其它的2,4-二氢-1,2,4-三唑-3-硫酮类化合物G11。以88%收率制备了5-[2-(8-氯-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)乙基]-4-甲基-2,4-二氢-1,2,4-三唑-3-硫酮(G11-b),mp216-219℃(EtOH)。1HNMR(DMSO-d6):2.41(3H,s),2.70(4H,s),3.06(2H,t),3.20(3H,s),3.49(2H,s),4.42(2H,t),7.05(1H,d),7.30-7.47(2H,m),13.55(1H,brs);MS,m/z361(M+)。以83%收率制备了5-[2-(8-氟-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)乙基]-4-甲基-2,4-二氢-1,2,4-三唑-3-硫酮(G11-c),mp215-218℃(EtOH)。1HNMR(DMSO-D6/CDCl3,2:1):2.43(3H,s),2.71(4H,s),3.04(2H,t),3.16(3H,s),3.48(2Hs),4.41(2H,t),6.83(1H,td),7.03(1H,dd),7.29(1H,dd),13.52(1H,brs);MS,m/z345(M+)。以93%收率制备了5-[2-(8-甲氧基-2-甲基-2,3,4,5-四氢-1H-吡啶并[4,3-b]吲哚-5-基)乙基]-4-甲基-2,4-二氢-1,2,4-三唑-3-硫酮(G11-d),mp219-222℃(EtOH)。1HNMR(DMSO-d6):2.43(3H,s),2.69(4H,s),3.05(2H,t),3.17(3H,s),3.50(2H,s),3.77(3H,s),4.39(2H,t),6.72(1H,dd),6.89(1H,d),7.27(1H,d),13.49(1H,brs);MS,m/z357(M+)。依据实施例制备的化合物在表4中进一步详述。表4.合成数据实施例60.本发明化合物与组胺受体结合能力的测定为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,2mMMgCl2,100mMNaCl,250mM蔗糖)中的中国仓鼠卵巢(CHO)细胞中所表达的人类重组组胺H1受体(DeBackerMD等Biochem.Biophys.ResComm.197(3):1601,1993)。将本发明化合物与1.2nM[3H]美吡拉敏在25℃孵育180分钟。在1μM美吡拉敏存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]美吡拉敏。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表5中。为评价本发明化合物在放射性配体结合试验中的活性,使用在50mM磷酸盐缓冲液(pH7.4)中的中国仓鼠卵巢(CHO)K1细胞中所表达的人类重组组胺H2受体(RuatM.ProcNatlAcadSciUSA.87(5):1658,1990)。将本发明化合物与0.1nM[125I]Aminopotentidine在25℃孵育120分钟。在3μM硫替丁存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[125I]Aminopotentidine。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表5中。表5.结合数据实施例1B:本发明化合物与肾上腺素能受体结合能力的测定。肾上腺素能α1A为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl缓冲液,pH7.4,0.5mMEDTA)中的取自威斯塔鼠颌下腺的大鼠肾上腺素能α1A受体(Michel,A.D.等,Br.J.Pharmacol.98:883,1989)。将本发明化合物与0.25nM[3H]哌唑嗪(Prozosin)在25℃孵育60分钟。在10μM酚妥拉明存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]哌唑嗪。在1μM或更低浓度筛选本发明化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。某些化合物显示抑制至少约80%的特异性结合。肾上腺素能α1B为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl缓冲液,pH7.4,0.5mMEDTA)中的取自威斯塔鼠肝的大鼠肾上腺素能α1B受体(Garcia-S'ainz,J.A.等,Biochem.Biophys.Res.Commun.186:760,1992;MichelA.D.等,Br.J.Pharmacol.98:883,1989)。将本发明化合物与0.25nM[3H]哌唑嗪在25℃孵育60分钟。在10μM酚妥拉明存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]哌唑嗪。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。某些化合物显示抑制至少约80%的特异性结合。肾上腺素能α1D为评价本发明化合物在放射性配体结合试验中的活性,使用在50mMTris-HCl缓冲液(pH7.4)中的人胚肾(HEK-293)细胞中所表达的人类重组肾上腺素能α1D受体(Kenny,B.A.等Br.J.Pharmacol.115(6):981,1995)。将本发明化合物与0.6nM[3H]哌唑嗪在25℃孵育60分钟。在10μM酚妥拉明存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]哌唑嗪。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。肾上腺素能α2A为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,12.5mMMgCl2,2mMEDTA)中的昆虫Sf9细胞中所表达的人类重组肾上腺素能α2A受体(UhlenS等JPharmacolExpTher.271:1558,1994)。将本发明化合物与1nM[3H]MK-912在25℃孵育60分钟。MK912是(2S-顺式)-1,3,4,5',6,6',7,12b-八氢-1',3'-二甲基-螺[2H-苯并呋喃并[2,3-a]喹嗪-2,4'(1'Η)-嘧啶]-2'(3'H)-酮盐酸盐。在10μMWB-4101(2-(2,6-二甲氧基苯氧基乙基)氨基甲基-1,4-苯并二烷盐酸盐)存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]MK-912。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。肾上腺素能α2B为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,12.5mMMgCl2,1mMEDTA,0.2%BSA)中的中国仓鼠卵巢(CHO-K1)细胞中所表达的人类重组肾上腺素能α2B受体(UhlenS等EurJPharmacol.343(1):93,1998)。将本发明化合物与2.5nM[3H]萝芙辛在25℃孵育60分钟。在10μM哌唑嗪存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]萝芙辛。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。肾上腺素能α2C为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,12.5mMMgCl2,2mMEDTA)中的昆虫Sf9细胞中所表达的人类重组肾上腺素能α2C受体(UhlenS等JPharmacolExpTher.271:1558,1994)。将本发明化合物与1nM[3H]MK-912在25℃孵育60分钟。在10μMWB-4101存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]MK-912。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。某些化合物显示抑制至少约80%的特异性结合。实施例2B:本发明化合物与多巴胺受体结合能力的测定。多巴胺D2L为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,1.4mM抗坏血酸,0.001%BSA,150mMNaCl)中的中国仓鼠卵巢(CHO)细胞中所表达的人类重组多巴胺D2L受体(Grandy,D.K.等Proc.Natl.Acad.Sci.USA.86:9762,1989;Hayes,G.等,Mol.Endocrinol.6:920,1992)。将本发明化合物与0.16nM[3H]螺哌隆在25℃孵育120分钟。在10μM氟哌啶醇存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]螺哌隆。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。实施例3B:本发明化合物与血清素受体结合能力的测定。血清素(5-羟基色胺)5-HT1A为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,0.1%抗坏血酸,0.5mMEDTA,10mMMgSO4)中的中国仓鼠卵巢(CHO-K1)细胞所表达的人类重组血清素(5-羟基色胺)5-HTIA受体(MartinGR和HumphreyPPA.Neuropharmacol.33:261,1994;MayJA,等JPharmacolExpTher.306(1):301,2003)。将本发明化合物与1.5nM[3H]8-OH-DPAT在25℃孵育60分钟。在10μM甲麦角林存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]8-OH-DPAT。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。血清素(5-羟基色胺)5-HT1B为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,154mMNaCl,10μM帕吉林,30μM异丙肾上腺素)中的来自威斯塔鼠大脑皮质的血清素(5-羟基色胺)5-HT1B受体(Hoyer等EurJPharmaco.118:1,1985;Pazos等EurJPharmacol.106:531,1985)。将本发明化合物与10pM[125I]氰基吲哚洛尔(Cyanopindolol)在37℃孵育90分钟。在10μM血清素(5-HT)存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[125I]氰基吲哚洛尔。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。血清素(5-羟基色胺)5-HT2A为评价本发明化合物在放射性配体结合试验中的活性,使用在50mMTris-HCl缓冲液(pH7.4)中的中国仓鼠卵巢(CHO-K1)细胞所表达的人类重组血清素(5-羟基色胺)5-HT2A受体(Bonhaus,D.W.等Br.J.Pharmacol.115:622,1995;Saucier,C.和Albert,P.R.,J.Neurochem.68:1998,1997)。将本发明化合物与0.5nM[3H]酮色林在25℃孵育60分钟。在1μM米安色林存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]酮色林。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。血清素(5-羟基色胺)5-HT2B为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,4mMCaCl2,0.1%抗坏血酸)中的中国仓鼠卵巢(CHO-K1)细胞所表达的人类重组血清素(5-羟基色胺)5-HT2B受体(Bonhaus,D.W.等,Br.J.Pharmacol.115:622,1995)。将本发明化合物与1.2nM[3H]麦角酸二乙酰胺(LSD)在37℃孵育60分钟。在10μM血清素(5-HT)存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]LSD。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。某些化合物显示抑制至少约80%的特异性结合。血清素(5-羟基色胺)5-HT2C为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,0.1%抗坏血酸,10μM帕吉林)中的中国仓鼠卵巢(CHO-K1)细胞所表达的人类重组血清素(5-羟基色胺)5-HT2C受体(Wolf,W.A.和Schutz,J.S.,J.Neurochem.69:1449,1997)。将本发明化合物与1nM[3H]美舒麦角在25℃孵育60分钟。在1μM米安色林存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]美舒麦角。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。血清素(5-羟基色胺)5-HT3为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,1mMEDTA,5mMMgCl2)中的人胚肾(HEK-293)细胞中所表达的人类重组血清素(5-羟基色胺)5-HT3受体(MillerK等Synapase.11:58,1992;BoessFG等Neuropharmacology.36:637,1997)。将本发明化合物与0.69nM[3H]GR-65630在25℃孵育60分钟。在10μMMDL-72222存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]GR-65630。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。某些化合物显示抑制至少约80%的特异性结合。血清素(5-羟基色胺)5-HT4为评价本发明化合物在放射性配体结合试验中的活性,使用在50mMTris-HCl(pH7.4)中的来源于DuncanHartley豚鼠纹状体的血清素(5-羟基色胺)5-HT4受体(GrossmanCJ等BrJPharmacol.109:618,1993)。将本发明化合物与0.7nM[3H]GR-113808在25℃孵育30分钟。在30μM血清素(5-HT)存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]GR-113808。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。血清素(5-羟基色胺)5-HT5A为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,10mMMgCl2,0.5mMEDTA)中的中国仓鼠卵巢(CHO-K1)细胞所表达的人类重组血清素(5-羟基色胺)5-HT5A受体(Rees,S.等,FEBSLett.355:242,1994)。将本发明化合物与1.7nM[3H]麦角酸二乙酰胺(LSD)在37℃孵育60分钟。在100μM血清素(5-HT)存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]LSD。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。某些化合物显示抑制至少约80%的特异性结合。血清素(5-羟基色胺)5-HT6为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,150mMNaCl,2mM抗坏血酸,0.001%BSA)中的人海拉(HeLa)细胞中所表达的人类重组血清素(5-羟基色胺)5-HT6受体(Monsma,FJ.Jr.等,Mol.Pharmacol.43:320,1993)。将本发明化合物与1.5nM[3H]麦角酸二乙酰胺(LSD)在37℃孵育120分钟。在5μM血清素(5-HT)存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]LSD。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。血清素(5-羟基色胺)5-HT7为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,10mMMgCl2,0.5mMEDTA)中的在中国仓鼠卵巢(CHO)细胞中表达的人类重组血清素(5-羟基色胺)5-HT7受体(Roth,B.L.等,J.Pharmacol.Exp。Ther.268:1403,1994;Shen,Y.等,J.Biol.Chem.268:18200,1993)。将本发明化合物与5.5nM[3H]麦角酸二乙酰胺(LSD)在25℃孵育2小时。在10μM血清素(5-HT)存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]LSD。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。实施例4B:本发明化合物与咪唑啉I2受体结合能力的测定。中枢咪唑啉I2为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl缓冲液,pH7.4,0.5mMEDTA)中的得自威斯塔鼠大脑皮质的大鼠中枢咪唑啉I2受体(Brown,CM.等,Br.J.Pharmacol.99:803,1990)。将本发明化合物与2nM[3H]咪唑克生在25℃孵育30分钟。在1μM咪唑克生存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]咪唑克生。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。某些化合物显示抑制至少约80%的特异性结合。实施例5B:本发明化合物与组胺受体结合能力的测定。组胺H1为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,2mMMgCl2,100mMNaCl,250mM蔗糖)中的在中国仓鼠卵巢(CHO)细胞中表达的人类重组组胺H1受体(DeBacker,M.D.等,Biochem.Biophys.Res.Comm.197(3):1601,1993)。将本发明化合物与1.2nM[3H]美吡拉敏在25℃孵育180分钟。在1μM美吡拉敏存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]美吡拉敏。在1μM筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示列于表6中。组胺H2为评价本发明化合物在放射性配体结合试验中的活性,使用在50mM磷酸盐缓冲液(pH7.4)中的中国仓鼠卵巢(CHO)K1细胞中所表达的人类重组组胺H2受体(Ruat,M.,Proc.Natl.Acad.Sci.USA.87(5):1658,1990)。将本发明化合物与0.1nM[125I]Aminopotentidine在25℃孵育120分钟。在3μM硫替丁存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[125I]Aminopotentidine。在1μM筛选化合物,使用1%DMSO作为溶媒。生物化学试验结果以特异性结合的抑制百分比表示于表6中。组胺H3为评价本发明化合物在放射性配体结合试验中的活性,使用在改良的Tris-HCl缓冲液(50mMTris-HCl,pH7.4,5mMMgCl2,0.04%BSA)中的中国仓鼠卵巢(CHO-K1)细胞所表达的人类重组组胺H3受体(YanaiK等JpnJPharmacol.65(2):107,1994;ZhuY等MolPharmacol.59(3):434,2001)。将本发明化合物与3nM[3H]R(-)-α-甲基组胺在25℃孵育90分钟。在1μMR(-)-α-甲基组胺存在下评价非特异性结合。过滤受体蛋白质,并洗涤,然后对过滤物计数以测定特异性结合的[3H]R(-)-α-甲基组胺。在1μM或更低浓度筛选化合物,使用1%DMSO作为溶媒。在该生物化学试验中测试了本发明化合物,且测定了特异性结合的抑制百分比。某些化合物显示抑制至少约80%的结合。表6:本发明化合物对配体与胺能G蛋白偶联受体结合的抑制:a在1nM的抑制百分比实施例6B:本发明化合物的血清素(5-羟基色胺)5-HT2A激动剂/拮抗剂活性的测定为测定本发明化合物在功能性试验中的激动剂或拮抗剂活性,使用人胚肾(HEK-293)细胞中所表达的人类重组血清素5-HT2A受体(JermanJC,BroughSJ,GagerT,WoodM,ColdwellMC,SmartD和MiddlemissDN.EurJPharmacol,414:23-30,2001)。将细胞混悬于DMEM缓冲液中,并分配于微孔板中。将胞浆钙荧光指示剂(随游离胞质Ca2+离子浓度成比例变化)与在补充有20mMHepes(羟乙基哌嗪乙磺酸)的HBSS缓冲液(pH7.4)中的丙磺舒(probenicid)混合,将其加入每个孔中,并在37℃与细胞平衡30分钟,随后在22℃平衡30分钟。为测定激动剂效应,将本发明化合物、参比激动剂或HBSS缓冲液(基础对照)加入细胞中,并使用酶标仪测定荧光强度的变化。对刺激对照的测定而言,将100nM5-HT加入单独的试验孔中。各结果以对100nM5-HT的对照响应的百分比来表示。标准参比激动剂是5-HT,在每个试验中对其的若干浓度进行测试,以得到浓度-响应曲线,从该曲线可以计算其EC50值。为测定拮抗剂效应,在萤光测定之前,加入本发明化合物、参比拮抗剂或HBSS缓冲液,随后加入3nM5-HT或HBSS缓冲液(基础对照)。各结果以表7中的对3nM5-HT的对照响应的抑制百分比来表示。标准的参比拮抗剂是酮色林,在每个试验中对其的若干浓度进行测试,以得到浓度-响应曲线,从该曲线可以计算其IC50值。在3μM或更低浓度筛选化合物,使用DMSO作为溶媒。表7:血清素5-HT2A拮抗剂试验血清素5HT2A拮抗剂试验实施例7B:本发明化合物的血清素(5-羟基色胺)5-HT6激动剂/拮抗剂活性的测定为测定本发明化合物在功能性试验中的激动剂或拮抗剂活性,将人类重组5-HT6受体转染于CHO细胞(Kohen,R.,Metcalf,M.A.,Khan,N.,Druck,T.,Huebner,K.,Lachowicz,J.E.,Meltzer,H.Y.,Sibley,D.R.,Roth,B.L.和Hamblin,M.W.Cloning,characterisationandchromosomallocalizationofahuman5-HT6serotoninreceptor(人5-HT6血清素受体的克隆、表征和染色体定位),J.Neurochem.,66:47,1996),并通过使用均相时间分辨荧光(HTRF)测定方法测定本发明化合物在cAMP产生中的作用以测定本发明化合物的活性。将细胞混悬于补充有20mMHEPES和500μMIBMX的HBSS缓冲液(pH7.4)中,然后在本发明化合物或参比激动剂或拮抗剂不存在(对照)或存在下,分配于微孔板中,并在37℃孵育45分钟。对于激动剂测定,在刺激对照的测定中,单独的试验测试孔中含有10μM5-HT。孵育后,将细胞裂解,并加入萤光受体(D2-标记的cAMP)和萤光供体(铕穴状化合物标记的抗-cAMP抗体)。在室温60分钟后,使用酶标仪在lex=337nm和lem=620和665nm测定荧光转移。通过用在665nm测定的信号除以在620nm测定的信号(比率)测定cAMP浓度。各结果以对10μM5-HT的对照响应的百分比来表示。标准的参比激动剂是5-HT,在每个试验中对其的若干浓度进行测试,以得到浓度-响应曲线,从该曲线可以计算其EC50值。对于拮抗剂测定,以100nM的最终浓度加入参比激动剂5-HT。对于基础对照测定,单独的试验孔中不包含5-HT。在37℃孵育45分钟后,将细胞裂解,并加入荧光受体(D2-标记的cAMP)和萤光供体(铕穴状化合物标记的抗-cAMP抗体)。在室温60分钟后,如以上所述测定荧光转移。各结果以对100nM5-HT的对照响应的抑制百分比来表示。标准的参比拮抗剂是美赛西平。实施例8B:化合物的多巴胺D2L拮抗剂活性的测定为测定本发明化合物在功能性试验中的激动剂或拮抗剂活性,使用在中国仓鼠卵巢(CHO)细胞中稳定表达的人类重组多巴胺D2L受体(SenoglesSE等JBiolChem.265(8):4507,1990)。将本发明化合物与在改良的HEPES缓冲液(20mMHEPES,pH7.4,100mMNaCl,10mMMgCl2,1mMDTT,1mMEDTA)中的膜(0.1mg/ml)和10mMGDP预孵育20分钟,并加入闪烁亲近试验(SPA)小珠在30℃再孵育60分钟。通过0.3nM[35S]GTPγS起始反应,再孵育15分钟。相对于1mM多巴胺的响应,本发明化合物增加50%或更多(350%)的[35S]GTPγS结合,表明其具有多巴胺D2L受体激动剂活性的可能性。本发明化合物对10μM多巴胺-诱导的[35S]GTPγS结合响应的增加的抑制达50%或更多(350%),表明了其具有受体拮抗剂活性。在3μM或更低浓度筛选化合物,使用0.4%DMSO作为溶媒。试验结果以特异性结合响应的百分比表示列于表8中。表8:本发明化合物的多巴胺D2L拮抗剂活性拮抗剂活性,以%抑制测定当列出两个值时,这表明试验进行了两次。实施例9B:本发明化合物的多巴胺D2S拮抗剂活性为测定本发明化合物在功能性试验中的激动剂或拮抗剂活性,使用在中国仓鼠卵巢(CHO)细胞中稳定表达的人类重组多巴胺D2S受体(GillilandSL和AlperRH.Naunyn-Schmiedeberg'sArchivesofPharmacology.361:498,2000)。将本发明化合物与在改良的HEPES缓冲液(20mMHEPES,pH7.4,100mMNaCl,10mMMgCl2,1mMDTT,1mMEDTA)中的膜(0.05mg/ml)和3μMGDP预孵育20分钟,然后加入闪烁亲近试验(SPA)小珠在30℃再孵育60分钟。通过0.3nM[35S]GTPγS起始反应,再孵育30分钟。相对于100μM多巴胺的响应,本发明化合物增加50%或更多(350%)的[35S]GTPγS结合,表明其具有多巴胺D2S受体激动剂活性的可能性。本发明化合物对3μM多巴胺-诱导的[35S]GTPγS结合响应的增加的抑制达50%或更多(350%),表明了其具有受体拮抗剂活性。在3μM或更低浓度筛选化合物,使用0.4%DMSO为溶媒。试验结果以特异性结合响应的百分比表示列于表9中。表9:本发明化合物的多巴胺D2S拮抗剂活性拮抗剂活性,以%抑制测定当列出两个值时,这表明试验进行了两次。实施例10B:在组胺H1功能性试验中对本发明化合物激动剂或拮抗剂活性的测定为测定本发明化合物在功能性试验中的激动剂或拮抗剂活性,使用人胚肾(HEK-293)细胞中所表达的人类重组组胺H1受体(Miller,T.R.,Witte,D.G.,Ireland,L.M.,Kang,C.H.,Roch,J.M.,Masters,J.N.,Esbenshade,T.A和Hancock,A.A.J.Biomol.Screen.,4:249-258,1999)。将细胞混悬于DMEM缓冲液中,然后分配于微孔板中。将胞浆钙荧光指示剂(随游离胞质Ca2+离子浓度成比例变化)与在补充有20mMHepes的HBSS缓冲液(pH7.4)中的丙磺舒(probenicid)混合,然后将其加入每个孔中,并在37℃与细胞平衡30分钟,随后在22℃再平衡30分钟。为测定激动剂效应,将本发明化合物、参比激动剂或HBSS缓冲液(基础对照)加入细胞中,并使用酶标仪测定荧光强度的变化。对刺激对照的测定而言,将10μM组胺加入单独的试验孔中。各结果以对10μM组胺的对照响应的百分比来表示。标准参比激动剂是组胺,在每个试验中对其的若干浓度进行测试,以得到浓度-响应曲线,从该曲线可以计算其EC50值。为测定拮抗剂效应,在萤光测定之前,加入本发明化合物、参比拮抗剂或HBSS缓冲液,随后加入300nM组胺或HBSS缓冲液(基础对照)。各结果以对300nM组胺的对照响应的抑制百分比来表示。标准的参比拮抗剂是酮色林,在每个试验中对其的若干浓度进行测试,以得到浓度-响应曲线,从该曲线可以计算其IC50值。在3μM或更低浓度筛选化合物,使用DMSO作为溶媒。实施例11B:与本发明化合物一起培养的神经元的神经突向外生长的增加皮质神经元中的神经突向外生长测试化合物以测定其刺激皮质神经元神经突向外生长的能力。使用标准方法分离皮质神经元。为分离原代大鼠皮质神经元,在Leibovitz培养基(L15;Gibco)中制备来自怀孕期第17天的怀孕大鼠的胚胎大脑。解剖出皮质,并去除脑脊膜。使用胰蛋白酶(Gibco)离解皮质C,并使用DNA酶I。用吸管在含有10%胎牛血清(“FBS”)(Gibco)的达尔伯克改良伊格尔培养基(“DMEM”;Gibco)中吹打分离细胞30分钟,并在室温以350xg离心10分钟。将细胞混悬于补充有2%B27(Gibco)和0.5mML-谷氨酰胺(Gibco)的Neurobasal培养基中。在37℃、5%CO2-95%空气的气氛下将细胞以30,000细胞/孔在涂覆有聚-L-赖氨酸的板上保存。贴壁后,向培养基中加入介质对照和在不同浓度的化合物。将BDNF(50ng/mL)用作神经突生长的阳性对照。处理后,将培养物在磷酸盐缓冲盐水(“PBS”;Gibco)中洗涤,并将其在2.5%在PBS中的戊二醛中固定。生长3天后细胞被固定。用照相机拍摄每种情况的带有神经突的细胞的一些图片(约80)。使用来自Image-ProPlus(法国)的软件分析所述图片以测定长度。结果以平均值表示(s.e.m.)。使用单向方差分析法(ANOVA)进行数据的统计分析。化合物395、68、250和20的结果在图1A-D中显示。在大鼠混合皮质培养物中的神经突向外生长由E18威斯塔鼠胚胎制备皮质混合培养物。解剖出皮质,并将组织切分为小份。用DNA酶和木瓜蛋白酶通过15分钟的孵育将细胞分离。通过离心(1500rpm,5分钟)收集细胞。用吸管吹打分离该组织,并使用micro-islet方案(在25μl介质中的20000个细胞)将细胞铺于聚-L-赖氨酸涂覆的48孔,其在补充有2mM谷氨酰胺、0.1μg/ml庆大霉素、10%热灭活的胎牛血清(FBS-HI)和10%热灭活的马血清(HS-HI)的MEM中。细胞附着于孔后,向各孔中加入250μl培养基。铺细胞4小时后,将培养基换为包含0.5、5和50nM浓度测试化合物的新鲜的培养基(含有补充物和5%HS-HI的MEM)。使用BDNF(50、100和/或150ng/ml)和/或NGF(50ng/ml和/或100ng/ml)作为阳性对照。体外研究2天后,在细胞固定前从板中收集细胞的条件培养基。将培养基样品在13000rpm离心以3分钟以除去细胞碎片。将样本在-20℃储存以用于之后的分析。用甲醛固定细胞,并为免疫细胞化学进行操作。使用制造商(Promega,BDNFImmunoAssaySystem,目录号:G7610)说明书用BDNFELISA测定条件培养基中的BDNF水平。将培养物用0.01MPBS中的4%甲醛固定30分钟,并用PBS洗涤一次。首先使固定的细胞透化,并使用含在PBS中的1%牛血清白蛋白和0.3%TritonX-100(聚乙二醇辛基苯基醚)的封闭缓冲液通过30分钟的孵育阻断非特异结合。将兔抗-MAP-2(稀释1:1000,AB5622,Chemicon,在封闭缓冲液中)用作第一抗体。将细胞与第一抗体在+4℃孵育48小时,用PBS洗涤,并与第二抗体——缀合于AlexaFluor568(1:200,A11036,分子探针)的羊抗-兔IgG在室温孵育2小时。通过配有适合的滤板系统的荧光显微镜目测检验免疫阳性细胞,并通过高分辨率图像捕获来记录。对每个区域的细胞(每孔4个区域)计数,并使用ImageProPlus软件定量测定神经突向外生长。每个浓度的化合物所使用的孔的数目是6(n=6)。所有数据以平均值±标准差(SD)或均值标准误(SEM)表示,且在p<0.05的水平时差异被认为是统计学上显著的。使用StatsDirect统计软件进行统计分析。通过使用单向-ANOVA、随后使用Dunnet检验(与介质处理组对比)分析各组平均值之间的差异。化合物395的结果在图1E中显示。实施例12B:使用体内模型在东莨菪碱处理的动物中评价化合物增强认知、学习和记忆的能力使用Ennaceur与Delacour开发的在大鼠中的双试验物体识别模型作为情景记忆的模型(Ennaceur,A.和Delacour,J.(1988),Behav.BrainRes.31:47-59)。该模型基于啮齿类自发的探究活动,且不涉及规则学习或强化。物体识别模型对老化和胆碱能功能障碍的效应敏感。参见,例如,Scali,C,等,(1994),Neurosci.Letts.170:117-120;和Bartolini,L.,等,(1996),Biochem.Behav.53:277-283。6至7周龄、重220-300克之间的雄性Sprague-Dawley大鼠来自Centred’Elevage公司(RueJanvier,B.P.55,LeGenest-Saint-Isle53940,法国)。将动物分为2-4组在以下标准条件下置于聚丙烯笼(地面面积1032cm2)中:在室温(22±2℃),在12小时亮/12小时暗的循环中,不限制食物和水。在治疗开始前允许动物适应环境条件至少5天,并在尾部用长久标记编号。试验场地是一个染为深蓝色的方形的木箱(60cmx60cmx40cm),在透明的有机玻璃地面下有一个15cmx15cm的黑色方形部分。每次试验之间用水清洁所述场地和放置在场地内的物体以除去任何大鼠留下的嗅迹。将场地置于一个暗室中,只用卤素灯泡朝向天花板照明,以在箱内获得均匀的大约60勒克司的暗光。测试前一天,在两个物体的存在下让动物自由探究试验场地3分钟(习惯化)。测试前将待测试动物放置于实验的房间中至少30分钟。在试验当天,对动物进行两次试验,间隔时间为120分钟。在第一次或获知试验(T1)期间,将大鼠置于有两个相同的物体的所述场地。测定每个动物完成15秒物体探究所需要的时间,停止时间为4分钟。鼻子距离物体的距离小于2厘米(“cm”)或接触物体被认为是探究。在第二次试验或测试试验(T2)期间,将第一次试验中存在的物体中的一个替换为未知的或新的物体,而第二个熟悉的物体放在原地。将大鼠放回场地3分钟,并测定对两个物体的探究。T1和T2期间对大鼠的运动行为(在透明的有机玻璃地面之下观察到的大鼠穿格次数)评分。试验最后,通过腹膜内施用过量的戊巴比妥处死大鼠。测定以下参数(1)T1期间完成15秒物体探究的时间;(2)T1期间的运动行为(穿格次数);(3)T2期间积极探究熟悉的物体所花费的时间(T熟悉);(4)T2期间积极探究新物体所花费的时间(T新);和(5)T2期间的运动行为(穿格次数)。评价T2期间积极探究新物体所花费的时间与T2期间积极探究熟悉的物体所花费的时间的差别(ΔT新-T熟悉)。还得出了每个组中T新-T熟悉大于或等于5秒的动物的百分比;以优秀学习者的百分比来描述。没有达到物体探究最低水平的动物由于其具有天然低的自发探究水平而被排除在研究之外。因而,该研究只包括对物体探究了至少5秒(T新+T熟悉>5秒)的大鼠。将动物随机分配至14个组。将本发明化合物和对照品如下所述施用于动物组:每天使用纯化水或盐水作为介质以0.25mg/ml的浓度新鲜配制化合物的溶液。将多奈哌齐用作阳性对照,并同时施用每天新鲜配制的东莨菪碱的单一的盐水溶液(5mL/kg)。将购自Sigma化学公司(目录号S-1875;St.QuentinFallavier,法国)的东莨菪碱以0.06mg/mL的浓度溶于盐水中。在获知试验(T1)之前40分钟经腹膜内施用多奈哌齐或其介质和东莨菪碱。获知试验(T1)之前25分钟通过管饲法施用化合物或其介质,即施用东莨菪碱5分钟后。就经腹膜内施用的化合物而言,施用的体积是5ml/kg体重,就口服施用而言是10ml/kg。化合物225、68和395的识别得分和优秀学习者的百分比在图2A-F中显示。实施例13B:使用体内模型在PCP处理的动物中测定化合物治疗、预防精神分裂症和/或延缓其发作和/或发展的能力精神分裂症的体内模型可以被用于测定本文所述的化合物治疗和/或预防精神分裂症和/或延缓其发作和/或发展的能力。用于测试本文所述的一种或多种化合物治疗和/或预防精神分裂症和/或延缓其发作和/或发展的能力的一种实例性的模型使用苯环利定,将其施用于动物(例如,非-灵长类动物(大鼠)或灵长类动物(猴子)),导致类似于在那些在患有精神分裂症的人中所发现的功能障碍。参见Jentsch等,1997,Science277:953-955和Piercey等,1988,LifeSci.43(4):375-385)。在这种或其它的动物模型中可使用标准的实验方案。一种方案涉及PCP-诱导的活动过度。使用来自JacksonLaboratories(BarHarbor,缅因州)的雄性C57B1/6J小鼠。所获得的是6周龄的小鼠。领受后,将小鼠标记唯一的识别号码(尾部标记),并且在每个OPTI小鼠通风笼中以4只小鼠/笼分组。在剩余研究期间所有的动物保持分组。测试前使所有小鼠适应动物房间至少两周,接下来测试时其平均年龄为8周。在适应期间,有规律地检查小鼠,对其进行处理并称重以确保足够的健康和适应性。将动物保持在12/12的亮/暗循环中。室温保持在20至23℃,相对湿度保持在30%至70%。研究期间不限制食物和水。在每个测试中,动物在治疗组之间随机分配。旷场(OF)测试评价运动活性。旷场箱是有机玻璃的方形的空间(27.3x27.3x20.3cm;MedAssociatesInc.,StAlbans,VT),环绕有红外光束(16x16x16)以测定水平和垂直活动。该分析的配置是将旷场分为中心和周围区域。当小鼠移动破坏水平光束时测定行进路程,而当垂直光束破坏时测定站立活动。在测试前将小鼠(每个治疗组10至12只动物)置于活性试验房间中适应至少1小时。在每次测试时,测试8只动物。对小鼠施用介质(10%DMSO或5%PEG200和1%吐温80)、本发明化合物、氯氮平,并放置于OF箱中30分钟,随后对其注射水或PCP,并放回OF箱中60分钟。在每次OF测试期的最后,将OF箱彻底清洁。在PCP活动过度小鼠的精神分裂症模型中的化合物36将化合物36(测试的剂量包括:0.05、0.15、0.45、1.5、4.5、15、30)溶于5%PEG200、1%吐温80中,在PCP注射30分钟之前口服施用。将氯氮平(1mg/kg)溶于10%DMSO中,并在PCP注射之前30分钟经腹膜内施用。将PCP(5mg/kg)溶于无菌注射用水中,并经腹膜内施用。通过方差分析(ANOVA)分析数据,随后酌情用Fisher检验进行事后比较(post-hoccomparisons)。在PCP注射之前测试的最初30分钟测定基线活性。在PCP注射之后的60分钟期间测定PCP-诱导的活性。从最终分析中除去落于距离平均值2个标准差以上或以下的统计异常值。如果p<0.05,则认为效应显著。在图3A-F中显示对于化合物36而言的总行进路程和总站立。在PCP活动过度小鼠的精神分裂症模型中的化合物247除如下的治疗组之外方案如上文所述:所有的注射的剂量体积为10ml/kg。以下化合物用于该研究:将化合物247(0.05、0.15、0.45、1.5、4.5和15.0mg/kg)溶于磷酸盐缓冲盐水(PBS)中,并在PCP注射30分钟之前口服施用。将氯氮平(0.5和1.0mg/kg)溶于10%DMSO中,并在苯环利定(PCP)注射之前30分钟经腹膜内施用。将PCP(5.0mg/kg)溶于无菌注射用水中,并经腹膜内施用。在图4A-C中显示对于化合物247而言的总行进路程。实施例14B:使用体内模型在苯丙胺处理的动物中测定化合物治疗、预防精神分裂症和/或延缓其发作和/或发展的能力使用来自适合的供应商(例如,JacksonLaboratories,BarHarbor,缅因州)的雄性C57B1/6J小鼠。通常获得6周龄的小鼠。测试前使所有小鼠适应动物房间至少两周。在适应期间,有规律地检查小鼠,对其进行处理并称重以确保足够的健康和适应性。将动物保持在12/12的亮/暗循环中。室温保持在20至23℃,相对湿度保持在30%至70%。研究期间不限制食物和水。在每个测试中,动物在治疗组之间随机分配。旷场(OF)测试评价运动活性。旷场箱是有机玻璃的方形箱(例如,27.3x27.3x20.3cm;MedAssociatesInc.,StAlbans,VT),环绕有红外光束源(16x16x16)。配置所述的箱以将旷场分为中心和周围区域,并设置光电池光束以测定在OF箱的中心和周围的活动。由连贯的光束破坏来测定水平活动(行进的路程)和垂直活动(站立)。在测试当天,在治疗前将动物置于试验房间中适应至少1小时。对动物施用介质、氯氮平、测试化合物,并放置于OF中。记录将待测化合物施用于每只动物的时间。记录基线活性30分钟,之后小鼠接受苯丙胺(4mg/kg)或水,放回OF箱中达60-分钟。在每次旷场测试期的最后,将OF箱彻底清洁。通常每组中测试10至12只小鼠。测试化合物剂量范围通常为0.01mg/kg至50mg/kg。通过方差分析(ANOVA)分析数据,随后酌情进行事后比较。如果p<0.05,则认为效应显著。数据以平均值和平均值的标准误(s.e.m)表示。实施例15B-本发明化合物对谷氨酸-诱导的45Ca2+摄入到大鼠大脑皮质的突触小体内的作用的测定。方法使用标准Hajos方法由新生(9-10日龄)威斯塔鼠的大脑皮质取得突触小体。将大脑与10个体积的冷却的0.32M蔗糖以900rpm匀化。将匀浆以1500g离心10分钟。将得到的上清液以10000g离心20分钟。通过将突触小体P2-部分混悬于孵育缓冲液A中累积放射性标记,孵育缓冲液A具有以下组成:132mMNaCl、5mMKCl、5mMHEPES、10mM葡萄糖、pH7.4(蛋白质浓度大约1.5-2mg/ml)。在37℃将该混合物与200μM谷氨酸再孵育5分钟,然后经GF/B过滤器(Whatman,英国)过滤来停止45Ca2+摄入,随后用冷的缓冲液B(145mMKCl,10mMTris,5.4mMTrilonB,pH7.4)洗涤三次。通过液闪β-分析仪(fluidscintillationbeta-analyzer)(TriCarb,PerkinElmer)分析放射性探针。所有的测试在2-3个独立地实验中进行三个平行测定。通过计算用谷氨酸刺激期间和没有激动剂刺激期间放射性标记的差异评价45Ca2+摄入的量。结果以占对照测定(为100%)的百分比来表示。使用以下公式评价特定的45Ca2+摄入:K(43/21)=[(Ca4-Ca3)/(Ca2–Ca1]100%,其中Ca1–对照的45Ca2+摄入(没有谷氨酸和测试化合物),Ca2-谷氨酸-诱导45Ca2+摄入(只有谷氨酸),Ca3-在测试化合物存在下45Ca2+摄入(没有谷氨酸),Ca4-在谷氨酸和测试化合物存在下的45Ca2+摄入。使用学生氏t检验对结果进行统计分析。结果研究了Dimebon对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。Dimebon在0.25-10μM的浓度抑制45Ca2+摄入到突触小体内的。在Dimebon浓度为1μM时观测到了对45Ca2+摄入到突触小体内的最大抑制(40%)。在浓度为0.1μM和100μM时,Dimebon引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加(相应地为120.4%和132%)(表10,图5A)。研究了化合物C4-1对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。化合物C4-1在0.1-0.5μM的浓度抑制谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内约20%。随着C4-1浓度增加,谷氨酸-诱导的45Ca2+摄入到突触小体内增加。当C4-1浓度为5μM时,观测到谷氨酸-诱导的45Ca2+摄入到突触小体内的最大增加(对照的128%)(表10,图5A)。研究了化合物C4-4对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。化合物C4-4在0.5μM的浓度抑制谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内。在浓度为1μM时观测到对45Ca2+摄入的最大抑制(20.5%)。C4-4浓度增加(50-100μM)引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加。在C4-4浓度为50μM时观测到谷氨酸-诱导的45Ca2+摄入到突触小体内的最大增加(对照的137.5%)(表10,图5A)。研究了化合物C4-5对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。化合物C4-5在1-25μM的浓度抑制谷氨酸-诱导的45Ca2+摄入到突触小体内。在C4-5浓度为10μM时观测到最大抑制(40%)。在0.5-0.1μM的浓度和在50-100μM的浓度时,化合物C4-5引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加。在C4-5浓度为100μM和0.1μM时观测到谷氨酸-诱导的45Ca2+摄入到突触小体内的最大增加(相应为148.4%和136.4%)(表10,图5A)。表10.Dimebon、C4-1、C4-4和C4-5对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。研究了化合物C4-6对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。C4-6在5-10μM的浓度抑制谷氨酸-诱导的45Ca2+摄入到突触小体内。随着化合物C4-6浓度增加(50-100μM),化合物C4-6引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加。在浓度为100μM观测到谷氨酸-诱导的45Ca2+摄入到突触小体内的最大增加(对照的152.2%)(表11,图5B)。表11.C4-6、C4-7、C1-1、C1-5、C1-7对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。研究了化合物C4-7对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。在浓度为0.1-10μM时,C4-7不影响谷氨酸-诱导的45Ca2+摄入到突触小体内。在浓度为50-100μM时,化合物C4-7引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加(对照的约128%)(表11,图5B)。研究了化合物C1-1对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。在浓度为0.1-0.5μM时C1-1抑制谷氨酸-诱导的45Ca2+摄入到突触小体内(~13%)。随着化合物C1-1浓度增加(50-100μM),化合物C1-1引起谷氨酸-诱导的45Ca2+摄入到突触小体内的增加。在浓度为50μM时观测到谷氨酸-诱导的45Ca2+摄入到突触小体内的最大增加(对照的126.9%)(表11,图6A)。研究了化合物C1-5对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。化合物C1-5抑制谷氨酸-诱导的45Ca2+摄入到突触小体内:浓度为0.5μM时,抑制约22.5%。化合物C1-5在100μM的浓度引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加(对照的130%)(表11,图6A)。研究了化合物C1-7对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。化合物C1-7抑制谷氨酸-诱导的45Ca2+摄入到突触小体内:浓度为0.1-0.5μM时,抑制约27.3%。化合物C1-7在5-100μM的浓度时引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加。在浓度为100μM时观测到谷氨酸-诱导的45Ca2+摄入到突触小体内的最大增加(对照的139.8%)(表11,图6B)。研究了化合物C1-6对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。C1-6在5μM的浓度抑制谷氨酸-诱导的45Ca2+摄入到突触小体内(抑制约20%)。浓度为0.1-1μM和10-100μM时,化合物C1-6引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加。在浓度为0.5μM和100μM时观测到谷氨酸-诱导的45Ca2+摄入到突触小体内的最大增加(分别为对照的142%和140.8%)(表12,图6C)。研究了化合物C1-8对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。C1-8在1-5μM的浓度抑制谷氨酸-诱导的45Ca2+摄入到突触小体内(抑制约14%)。在浓度为50-100μM时化合物C1-8引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加(对照的约122%)(表12,图6D)。研究了化合物C1-4对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。化合物C1-4在0.5μM的浓度抑制谷氨酸-诱导的45Ca2+摄入到突触小体内(抑制约12.6%)。在浓度为50-100μM时,化合物C1-4引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加(对照的约131%)(表12,图6D)。研究了化合物C4-3对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。在浓度为0.1-50μM时,C4-3不影响谷氨酸-诱导的45Ca2+摄入到突触小体内。在浓度为50-100μM时,化合物C4-3引起谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的增加(对照的约126%)(表12,图7)。研究了化合物C2-1对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内的作用。在浓度为0.1-1μM时,C2-1不影响谷氨酸-诱导的45Ca2+摄入到突触小体内。在浓度为5-50μM时,化合物C2-1抑制谷氨酸-诱导的45Ca2+摄入到突触小体内。在浓度为10μM时,对谷氨酸-诱导的45Ca2+摄入到突触小体内的最大抑制为30%(表12,图7)。表12.化合物C1-6、C1-8、C1-4、C2-1对谷氨酸-诱导的45Ca2+摄入到大鼠皮质突触小体内中的作用。实施例16B-本发明化合物对AMPA-受体活性的作用的测定。方法使用电生理膜片钳方法在由新生小鼠(12-15日龄)的小脑中新鲜分离的浦肯野细胞的全细胞模式(Hamill等,1980)中分析测试化合物对AMPA受体的作用。使用改良的方法(Kaneda等,1988)进行分离,其中将大脑的400-600μm切片在温度控制箱在10mL缓冲溶液(150mMNaCl,5mMKCl,2mMCaCl2,2mMMgSO4·7H2O,10mMHEPES,15mM葡萄糖,pH7.42)中孵育60分钟。将孵育溶液与有链霉蛋白酶(2mg/mL)和胶原酶(1mg/mL)的孵育缓冲液交换70分钟。用初始的缓冲液洗涤20分钟后,将所有的切片置于培养皿中,并使用巴斯德吸管分离。在34℃用100%O2对所有的溶液持续充气。细胞外盐水的组成为:150mMNaCl、5mMKCl、2.6mMCaCl2、2mMMgSO4·7H2O、10mMHEPES、15mM葡萄糖,pH7.36。通过表面灌流激动剂溶液(红藻氨酸)活化AMPA受体,结果诱导跨膜电流。使用硼硅酸盐微电极(电阻2.5-4.0mOhm)测定电流,其中填充100mMKCl、11mMEGTA、1mMCaCl2、1mMMgCl2、10mMHEPES、5mMATP,pH7.2。使用EPC-9(HEKA,德国)记录电流,并用同样购自HEKA的Pulse软件包记录。使用Pulsefit(HEKA,德国)对所观测到的进行分析,并使用学生氏t-检验标准分析统计学显著性。数据以占对照值的百分比的平均值±SEM表示。结果在3-5个神经元中测试本发明化合物。每个浓度的化合物在每个神经元中重复3-4次。数据以占对照值的百分比的平均值±SEM(表13+14,图8-12)表示。对照值=100%。表13.新衍生物对红藻氨酸-诱导的浦肯野细胞中的电流的作用。化合物10-9M10-8M10-7M10-6M10-5M3x10-5MC4-1125±6147±11115±3110±597±4未测定C4-5100131±3133±5108±7106±6未测定C4-4100129±5147±15142±11150±26150±20C4-6未测定100±3120±20140±25152±30149±33C4-7未测定95±8118±12109±17106±14101±9C1-1100±3149±25148±1498±2105±895±6C1-564±862±1270±2364±23126±18未测定表14.C系列新衍生物对红藻氨酸-诱导浦肯野细胞中的电流的作用。化合物10-9M10-8M10-7M10-6M10-5M3x10-5MC1-6100±3101±4116±7146±12135±1130±6C1-4129±9119±6114±5109±12100±9未测定C1-8未测定100±4102±3123±8105±890±7C4-3100±3145±12149±15139±11100±680±10实施例17B-化合物对线粒体的影响方法由雄性威斯塔鼠制备大鼠肝和脑线粒体。使大鼠在CO2箱内安乐死,随后断头,该方法与IPACRAS的动物实验指南一致。在4℃经标准甘露醇差速离心方案分离在充足的缓冲液A中的来自大鼠肝的线粒体,所述缓冲液A包含210mM甘露醇、70mM蔗糖、5mMHEPES、0.25mMEDTApH7.4,并将其重混悬于没有EDTA的缓冲液A中用于mPT的研究。脑线粒体从混合的两只大鼠的前脑中分离。使用Sims的改良的方法分离并以Percoll梯度纯化脑线粒体。将所述脑在“缓冲液A”(225mM甘露醇,75mM蔗糖,5mMHEPES,1mMEGTA,pH7.4[KOH])中匀化,并以1300g离心10分钟。将上清液以10000g离心10分钟,然后将得到的沉淀混悬于15%的Percoll中,并铺于分别由40和23%的Percoll组成的不连续梯度之上,然后将其在30000g离心10分钟,离心过程中不制动。由较低两层的界面收集线粒体,在重悬于“缓冲液A”中后,将该混悬液以16600g离心10分钟,然后将得到的沉淀重悬于“缓冲液A”,加入BSA(10mg/mL)以结合游离脂肪酸;并将该混悬液以6300g再次离心10分钟。离心后将小球状的线粒体重悬于没有EGTA的分离培养基中以达到15-20mg/ml的蛋白质浓度。肿胀试验。由溶质经开放的PT孔道的流入所引起的线粒体肿胀导致光透过的增加(即,浊度降低)。通过测定线粒体混悬液中的吸光度,这种浊度变化提供了一个方便且经常使用的MPT的试验。在本研究中,由Ca2+和无机磷酸盐(Ca/Pi)诱导的MPT通过在BeckmanDU640分光光度计中的比色皿中的1ml的缓冲液A加0.8μM鱼藤酮、5mM琥珀酸盐、1mMKH2PO4和0.5mg或0.2mg的分离出的肝或脑的线粒体的蛋白质在540nm处的吸光度的变化而相应地监测。反应通过加入触发剂(Ca2+)开始。在30℃进行预处理和反应。肿胀速度被定量化为ΔA540/分钟/mg,其在所有情况中从A540对时间的图的最陡部分的切线计算。线粒体膜电位的测定。将相同的试验条件用于对线粒体膜电位变化的评价,不同之处是孵育培养基中包括最终浓度10mkM的番红且在本发明化合物加入之前加入了琥珀酸盐。由于最佳浓度能在信号/基线比率与番红自身对由Ca/Pi诱导的肿胀的干扰(在浓度超过20mkM番红有增强Ca/Pi–诱导的肿胀的倾向)之间取得最佳的折中结果,所以事先测定番红的该浓度。在BeckmanDU640分光光度计的比色皿中在554和524nm处以分光光度法测定来评价mPT孔道状态的变化。A554-524表示线粒体电位的特征且A554表示线粒体肿胀的特征。线粒体钙保留能力的测定。在供给能量的条件下,在BeckmanDU640分光光度计中的比色皿中的包含5mkM偶氮胂III、1μg/ml寡霉素和20μMADP以及作为呼吸底物的5mM苹果酸和谷氨酸的基于蔗糖的缓冲液(250mM蔗糖,10mMHEPES,1mMPi(K),1mMMgCl2,10μMEGTA,pH7.2)中,在推注单次剂量的Ca2+后分析线粒体外的Ca2+。结果使用Ca2+-诱导的线粒体肿胀作为化合物对MPT孔道开放影响的初步筛选试验。测定IC50(其为在加入钙后使浊度降低的初始速度降低多达50%的化合物浓度)。但肿胀的抑制可能不只与MPT孔道的打开的直接抑制相关,还可与线粒体的呼吸功能的损害以及化合物对钙转移至线粒体中的作用有关。为对这些可能性进行初步判断,测定线粒体膜电位和线粒体钙保留能力。依据实验数据(表15),其表明至少特别是C4-1、C4-4、C4-6和C1-5对MPT的抑制伴随有线粒体呼吸功能的损伤。新的合成的化合物(除C1-4和C4-3外)单独诱导脑线粒体悬液浊度的增加。这种作用的机制需要进一步的研究。表15.化合物对大鼠脑线粒体的功能特征的影响。化合物C1-6和C1-8减少钙-诱导的脑线粒体肿胀,IC50分别接近12μM和6μM,但当浓度更高时观测到作用降低(图13),加入100μMC1-8后观测到线粒体悬液吸光度显著提高(表15)。这两种化合物在接近IC50的浓度时增加线粒体的钙保留能力。在番红测试中,C-1-6和C-1-8显示出去极化活性,但这两种化合物在没有线粒体的缓冲液均不与番红反应。化合物C1-4和C4-3降低钙-诱导的脑线粒体肿胀,其IC50分别接近94μM和76μM(表15,图13)。在浓度高至100mkM时,没有观测到这些化合物引起脑线粒体去极化。实施例18B-化合物对空间记忆性能和在识别记忆任务(物体识别测试)中的性能的影响物体识别测试基于以下事实:小鼠和大鼠本能的探究新的物体或已知物体的新位置比已知的物体或物体的已知位置所用的时间长得多。该测试首先用于大鼠中(EnnaceurA.,DelacourJ.,1988)。随后通过其他人研究显示这种记忆测试适合于小鼠(DodartJ.等1997,MessierC.1997,PittengerC.等,2002,RyabininA.等,2002,SargoliniF.等,2003)。物体识别的方法被分为两个阶段。第一个阶段涉及识别已知物体的新的位置,其用于评价空间记忆。第二个阶段包括识别新物体的测试,以评估非空间记忆(GaffanD.,1992,KoIbB.等,1994,StecklerT.,1998)。目前,这两种都被广泛应用以测定新合成药物对记忆的效果。物质和方法所有的试验都在3-4个月龄大的雄性的(C57BL/6系)、重22-26g的动物中进行,每组10只动物。所有动物都被放在动物笼(每笼5只动物)中,保持12x12小时亮循环(上午8点-下午8),不限制食物和水。观测箱由不透明的白色塑料制成48x38x30cm(18.9x14.9x11.8英寸)。使用褐色玻璃小瓶作为测试物体,其直径为2.7cm(1.1英寸),高为5.5cm(2.2英寸)。在每次测试前用85%乙醇将所有的小瓶处理2-3分钟。将动物放置于箱的中央。将Dimebon和本发明化合物溶于蒸馏水中,并在训练练习前1小时以每10g动物质量0.05mL口服施用。对照组中的所有的动物用相等剂量的介质处理。训练练习48小时后进行测试,因为动物在48小时休息时间内遗忘物体的位置。实验过程已知物体的新的位置的识别(空间记忆性能)对测试箱的适应第一天将动物放入测试房间中以使其能对测试环境适应20-30分钟。随后,将每只动物放在事先用酒精擦拭的空的测试箱中10分钟。之后将动物放回笼中并返回动物房间中。训练练习适应后,在第二天,将动物放回测试房间中,对其进行称重并注射测试化合物。注射后1小时,将每只动物放入测试箱,箱中有放置于对角线上的距角落14.5cm(5.7英寸)的两个相同的物体。每只动物训练练习持续15分钟。接着将每只动物放回笼中并返回动物房间。测试过程在训练练习48小时后进行测试过程。这一过程期间如在训练练习中那样使用两个物体。一个物体留在原来的位置而另一个放在新的位置。使用两个电子计时表以0.1秒的精度测定每只动物在探究每个物体中所花费的时间,总共记录10分钟。由镜子观测动物。如果动物的鼻子距离物体2cm内或如果动物直接用鼻子接触物体则对物体的探究计为阳性。新物体的识别(对识别记忆任务的非空间的性能)以下试验也在3-4个月龄大的雄性的(C57BL/6系)的动物中进行,每组10只动物。所有动物都被放在动物笼(每笼5只动物)中,保持12x12小时亮循环(上午8点-下午8),不限制食物和水。然而,在此使用直径为52cm(20.5英寸)、高为34cm(13.4英寸)的圆底测试箱。同样,使用褐色玻璃小瓶作为测试物体,其直径为2.7cm(1.1英寸),高为5.5cm(2.2英寸)。将本发明化合物在训练练习前1小时以每10g动物质量0.05mL口服施用。对照组中的所有的动物用相等剂量的介质处理。训练练习48小时后进行测试,因为动物在48小时休息时间内遗忘物体的位置。试验过程与之前所述的测试(已知物体的新位置的识别)相同。主要的不同是在测试过程中有一个已知的物体(褐色玻璃小瓶)和一个未知的物体。第二个玻璃小瓶被金属圆筒取代,其直径3cm(1.2英寸),高4.5cm(1.8英寸)。统计数据分析由于每只动物探究物体所用时间很不同,使用以下公式测定每只动物探究时间的百分比:tNL/(tFL+tNL)x100(其中t是时间,NL–新的位置(物体)和FL–熟悉的位置(物体))。将动物探究两个物体的总的时间作为100%。随后使用学生氏t检验标准分析统计学显著性。结果在物体识别测试中Dimebon对空间和非-空间记忆的作用的比较评价1.新物体识别测试中Dimebon对记忆的作用在该实验中,在训练前将1小时将Dimebon以宽的剂量范围(从0.005mg/kg至2.0mg/kg)施用。结果发现在0.005mg/kg时,Dimebon具有记忆活化作用。所记录的产生最大作用的剂量是0.05mg/kg。在该组中动物花费总时间的65.6±4.4%识别新的物体,而花费总时间的34.4±4.4%识别已知的物体(P=0.0003)。暴露剂量的进一步增加导致刺激作用的降低。此外,在0.5mg/kg时实验的结果不是统计学显著的。然而,进一步增加暴露剂量高至1-2mg/kg时,对记忆再次产生刺激作用(图14)。因此,Dimebon显示对非空间记忆具有双相活化作用,这可能与其活化作用的至少两种不同的机制相关。2.在新的位置的已知物体的识别任务中Dimebon对记忆的作用。之前显示在识别新的位置的已知物体的测试中Dimebon在0.01-0.1mg/kg时对记忆具有剂量依赖性的活化作用。然而,当剂量0.25mg/kg时该作用消失,因而在Dimebon更高的暴露剂量时,测试其活化波是否出现第二个阶段是重要的。结果显示只有在10mg/kg时Dimebon对记忆具有显著的刺激作用。该组中的动物花费总时间的61.2±5%探究在新位置的已知的物体,且花费总时间的38.8±5%探究已知的位置(P=0.0003)(图15)。因此,可断定Dimebon对空间记忆也具有两个阶段的刺激作用(如在识别在新位置的已知物体的测试中所显示的);然而需要剂量高很多的该药物。在识别新位置的已知物体的测试中化合物对记忆的作用进行试验来评价对熟悉物体的新的位置的识别,以研究C4-1、C4-4、C4-5、C4-6、C4-7、C1-1和C1-5对记忆的作用。训练练习48小时后将对照动物约在同时探究在已知的和新的位置的物体。因此,它们忘记了训练期间的物体位置。然而,C4-1(0.1mg/kg)治疗的动物探究在新的位置物体花费总时间的61.3±6.9%,探究在已知位置的物体花费38.7±6.9%(P=0.0004)。其他组中的结果(0.05mg/kg、0.5mg/kg和1mg/kg)与对照组相比没有显著不同(图16)。因此,C4-1只在剂量为0.1mg/kg时对记忆具有活化作用。对C4-4的研究显示在剂量为0.05mg/kg时其对动物探究在已知或在新的位置的物体所用的时间没有作用。然而,用0.1mg/kgC4-4治疗的动物的组探究在新的位置的物体花费总时间的58.5±9.6%,且探究在已知位置的物体花费41.5±9.6%(P=0.01)。用0.5mg/kgC4-4治疗的小鼠探究在新的位置的物体花费总时间的59.1±4.2%,且探究在已知位置的物体花费40.9±4.2%(P=0.0003)。在用1mg/kgC4-4治疗的小鼠中得到相似的结果(图17)。C4-4对记忆具有活化作用,该作用在训练后约48小时仍然保留。该作用与C4-1的作用相似,但C4-4具有的有效剂量的范围更宽。在新位置的已知物体的识别测试中以0.05mg/kg、0.1mg/kg、0.5mg/kg和1mg/kg的剂量测试C4-5。0.5mg/kg组动物探究在新位置的物体花费总时间的56.2±7.6%,探究在已知位置的物体花费43.8±7.6%(P=0.03)。当剂量增加至1mg/kg,动物探究在新位置的物体花费总时间的60.6±8.4%,探究在已知位置的物体花费39.4±8.4%(P=0.006)。0.05mg/kg和0.1mg/kg剂量没有作用(图18)。因此,C4-5刺激记忆,但活性比C4-1和Dimebon中的一个弱约10倍。用0.5mg/kg剂量的化合物C4-6治疗的动物探究在新位置的物体花费总时间的61.0±4.8%,探究在已知位置的物体花费39.0±4.8%(P=0.0007)。当剂量增加至1mg/kg,动物探究在新位置的物体花费总时间的56.3±7.7%,探究在已知位置的物体花费43.7±7.7%(P=0.03)。0.05mg/kg和0.1mg/kg剂量没有作用(图19)。因此,C4-6刺激记忆,但活性比Dimebon中的一个弱约5倍。在新位置的已知物体的识别测试中以0.05mg/kg、0.1mg/kg、0.5mg/kg和1mg/kg的剂量测试C4-7。已测定0.1mg/kg组探究在新位置的物体花费总时间的63.4±10.4%,探究在已知位置的物体花费36.6±10.4%(P=0.01)。0.05、0.5和1mg/kg剂量没有作用(图20)。因此,C4-7刺激记忆,且具有与Dimebon相似的活性,但是有效剂量范围小很多。在Dimebon对空间记忆的两个阶段的作用的测试中发现了C1-1的一个相当令人感兴趣的记忆活化作用。注射了0.05mg/kg的C1-1的小鼠探究在新位置的物体花费总时间的61.8±8.4%,探究在已知位置的物体花费38.2±8.4%(P=0.002)。当剂量增加到高至0.1mg/kg,动物花费总时间的56.0±6.9%以确定在新位置的物质,且探究在已知位置的物体花费42.7±6.9%(P=0.02)。而且,进一步增加至0.5mg/kg时,导致对记忆刺激作用消失。然而,注射1mg/kg的C1-1组的小鼠探究在新位置的物体花费总时间的61.5±8.9%,探究在已知位置的物体花费37.8±8.9%(P=0.002)。换言之,再次观测到了刺激作用(图21)。C1-1对记忆具有刺激作用。该化合物的活性比Dimebon中的一个高约两倍,但相比之下C1-1具有两个阶段的响应作用。但是,C1-1出现第二个刺激阶段的浓度比对Dimebon而言所显示的浓度低约10倍。完全可能的是该化合物对空间记忆的刺激作用会比对于Dimebon而言所显示的作用高的多。对C1-5的研究显示在剂量0.05mg/kg时其对动物探究在已知或在新的位置的物体所用的时间没有作用。然而,用0.1mg/kgC1-5治疗的动物的组探究在新位置的物体花费总时间的57.9±7.5%,探究熟悉位置的物体花费42.1±7.5%(P=0.01)。用0.5mg/kgC1-5治疗导致对记忆刺激作用消失。然而,当剂量增加高至1mg/kg,动物探究在新位置的物体花费总时间的57.4±8.4%,探究熟悉位置的物体花费42.6±8.4%(P=0.03)。基于已得到的结果得出C1-5对记忆有刺激作用的结论,且其活性是在Dimebon的水平。如C1-1一样,该化合物也具有两个阶段的刺激作用(图22.)。结果在表16中概述。表16.在识别在新位置的已知物体的测试中多个剂量的C系列化合物对记忆的作用。+该剂量对记忆有活化作用-发现该剂量对记忆没有活化作用参考文献·DodartJ.C.,C.Mathis和A.Ungerer,Scopolamine-induceddeficitsinatwo-trialobjectrecognizetaskinmice(在小鼠中的两个试验物体识别任务中东莨菪碱所诱导的缺陷).NeuroReport8(1997),第1173-1178页.·EnnaceurA.和J.Delacour,Anewone-trialtestforneurobiologicalstudiesofmemoryinrats.1:behavioraldata(用于大鼠记忆的神经生物学研究的一种新的一个试验测试.1:行为数据).Behav.BrainRes.31(1988),第47-59页。·KoIbB.,K.Buhrmann,R.McDonalD和R.Sutherland,Dissociationofthemedialprefrontal,posteriorparietal,andposteriortemporalcortexforspatialnavigationandrecognitionmemoryintherat(在大鼠中分离内侧前额叶、顶叶后和后颞叶皮质用于空间导航和识别记忆).Cereb.Cortex6(1994),第664-680页。·MessierC,Objectrecognitioninmice:improvementofmemorybyglucose(小鼠中的物体识别:通过葡萄糖改善记忆).Neurobiol.Learn.Mem.67(1997),第172-175页。·A.E.Ryabinin,M.N.Miller和S.Durrant,EffectsofacutealcoholadministrationonobjectrecognitionlearninginC57BL/6Jmice(急性酒精施用对C57BL/6J小鼠物体识别学习的作用).Pharmacol.Biochem.Behav.71(2002),第307-312页。·SargoliniF.,P.Roullet,A.Oliverio和A.Mele,Effectsofintra-accumbensfocaladministrationsofglutamateantagonistsonobjectrecognitionmemoryinmice(伏隔核内局部施用谷氨酸拮抗剂对小鼠中物体识别记忆的作用).Behav.BrainRes.138(2003),第153-163页。·SikA.,vanNieuwehuyzen,J.Prickaerts,A.Blokland,Performanceofdifferentmousestrainsinanobjectrecognitiontask(在物体识别任务中不同小鼠品系的表现).Behav.BrainRes.147(2003),第49-54页。·StecklerT.,W.H.I.M.Drinkenburgh,A.Sahgal和J.P.Aggleton,Recognitionmemoryinrats.I.Conceptsandclassification(大鼠中的识别记忆I.概念和分类).Prog.Neurobiol.54(1998),第289-311页。·StecklerT.,W.H.I.M.Drinkenburgh,A.Sahgal和J.P.Aggleton,RecognitionmemoryinratsII.Neuroanatomicalsubstrates(大鼠中的识别记忆II.神经解剖学底物).Prog.Neurobiol.54(1998),第313-332页。应理解本文详述的实施方案,当可适用时,同等地应用于全文提供的式。还应理解本发明包括式(B)化合物,其中X7-X10,R1、R2a、R2b、R3a、R3b、R8c-R8f,R10a,R10b、m、q如在式(I)中的一个实施方案中和式(E)中的另一个实施方案所定义。全文的所有参考,诸如出版物、专利、专利申请和已公开的专利申请,被全部引入本文作为参考。尽管为清楚理解目的以上发明在某种程度上详细地通过例举和实施例的方式描述,但本领域熟练的技术人员显然可以进行某些小的变化和改进。因此,所做的说明和实例不应当被认为是对本发明范围的限制。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1