一种用于合成拉帕替尼或其中间体5‑(4‑羟基喹唑啉)‑呋喃‑2‑甲醛的方法与流程
本发明属于医药合成
技术领域:
,尤其是涉及拉帕替尼及其中间体的合成方法。
背景技术:
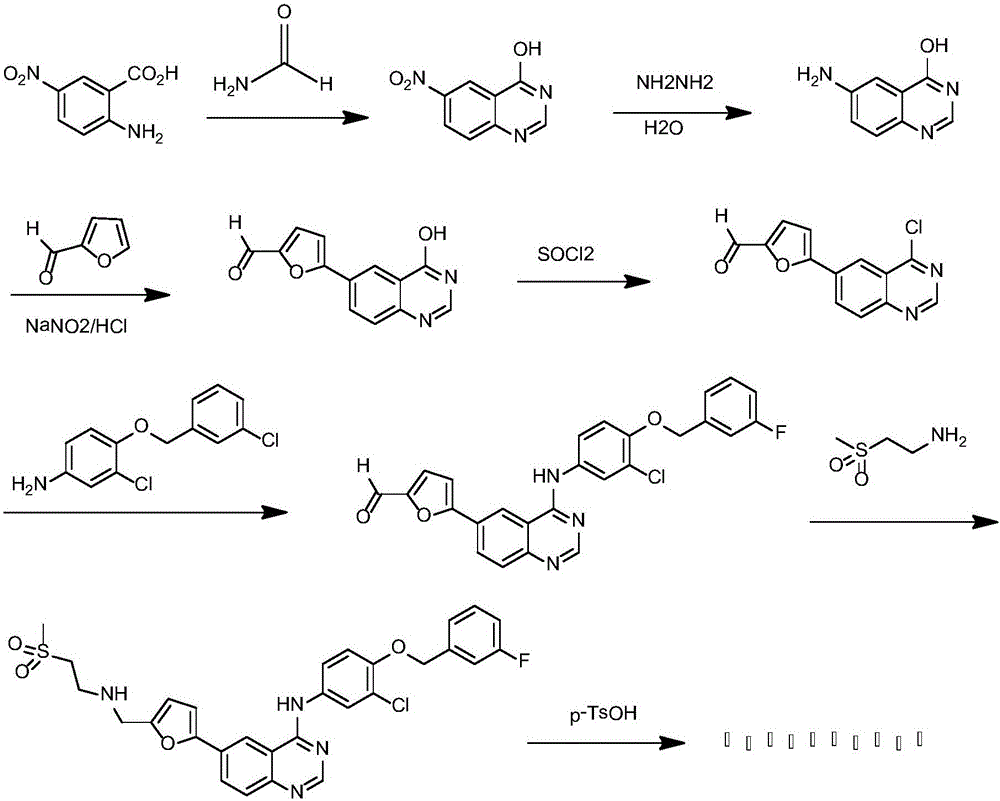
:拉帕替尼是小分子4-苯胺基喹唑啉类受体酪氨酸激酶抑制剂,抑制表皮生长因子受体(ErbB1)和人表皮因子受体2(ErbB2)。4种乳腺癌细胞株中BT474和SKBr3对拉帕替尼敏感,半抑制浓度为25和32nmol/L,MDA-MB-468和T47D细胞株不敏感,半抑制浓度在微摩尔级别级别,对于膀胱癌的2种细胞株,RT112(ErbB1和ErbB2高度表达)和J82(ErbB1和ErbB2低度表达),增强顺铂的疗效。在多种动物均能抑制表皮因子驱动的肿瘤生长。拉帕替尼对曲妥单抗耐药的肿瘤细胞株有效。现有关键中间体的合成方法采用价格昂贵的碘试剂作为原材料,在金属钯或其衍生物催化下通过Suzuki偶联反应与呋喃甲醛硼酸偶合。专利W09935146,W00104111,W00202552,W005046678,W008067144,等报道了拉帕替尼、拉帕替尼二对甲苯磺酸盐一水合物,及其相关中间体的制备方法。已知合成路线中,以6-卤代-喹唑啉-4-酮为原料,与氯代试剂如二氯亚砜(SOCl2)、三氯氧膦(POCl3)反应制备6-卤代-4-氯喹唑啉),再与3-氯-4-(3-氟苄氧基)苯胺经取代反应得中间体N-[3-氯-4-[(3-氟苄基)氧]苯基]-6-(溴)碘喹唑啉-4-胺。中间体N-[3-氯-4-[(3-氟苄基)氧]苯基]-6-碘喹唑啉-4-胺与糠醛衍生物通过过渡金属催化的偶联反应得5[4-({3_氯-4-[(3-氟苄基)氧]苯基}氨基)喹唑啉-6-基]-2-呋喃甲醛,再与2-甲磺酰乙胺盐酸盐或游离碱经还原胺化反应制备拉帕替尼。图1显示了现有技术中的一种拉帕替尼合成路线。然而,现有技术在合成拉帕替尼及其中间体的过程中还存在过程繁杂、反应条件/操作要求高、试剂昂贵且利用率低、重金属污染和/或产率低等问题,因此在帕替尼及其中间体的过程中,仍然迫切需要提供能够解决上述一个或者多个现有技术问题的合成方法。技术实现要素:为了解决拉帕替尼及其中间体合成过程中存在的一个或者多个上述问题,本发明在第一方面提供了一种制备拉帕替尼的中间体的方法,所述中间体为5-(4-羟基喹唑啉)-呋喃-2-甲醛,所述方法包括如下步骤:步骤(1):在溶剂中在催化量的催化剂存在的情况下,使4-羟基-6-硝基喹唑啉与水合肼反应,制得4-羟基-6-氨基喹唑啉;和步骤(2):在溶剂中在酸、亚硝酸钠和催化量的催化剂存在的情况下,使4-羟基-6-氨基喹唑啉与呋喃甲醛反应,制得所述中间体。本发明在第二方面还提供了一种合成拉帕替尼和/或其盐、拉帕替尼中间体和/或所述中间体的药学可接受的盐的方法,所述方法利用5-(4-羟基喹唑啉)-呋喃-2-甲醛进行,并且所述5-(4-羟基喹唑啉)-呋喃-2-甲醛由权利要求1至5中任一项所述的方法合成。本发明采用创新的合成路线及方法,合成拉帕替尼及其关键中间体。本发明方法简化现有技术中拉帕替尼及其关键中间体合成过程中涉及的合成步骤;所采用的原料便宜易得,例如现有技术中使用昂贵的特殊试剂如有机钯催化剂以及昂贵并且制备过程复杂的试剂如6-卤代-喹唑啉-4-酮、呋喃甲醛硼酸,相反,本发明方法所使用的大部分所需试剂如2-氨基-5-硝基-苯甲酸和呋喃甲醛等都已经商品化,使用了以结构简单、价格便宜、不涉及重金属残留的无机铜试剂为主的催化剂来代替现有技术中以有机钯催化剂为主的昂贵催化剂,从而降低原料和试剂成本,避免现有合成方法存在的如原材料成本较贵的问题;本发明方法还克服了现有技术中反应条件/操作要求高、试剂利用度低和/或产率低等问题。附图说明图1显示了现有技术中的一种拉帕替尼合成路线。图2显示了使用本发明方法合成拉帕替尼及其关键中间体的路线。具体实施方式如上所述,本发明在第一方面提供了一种制备拉帕替尼的中间体的方法,所述中间体为5-(4-羟基喹唑啉)-呋喃-2-甲醛,所述方法包括如下步骤:步骤(1):在溶剂中在催化量的催化剂存在的情况下,使4-羟基-6-硝基喹唑啉与水合肼反应,制得4-羟基-6-氨基喹唑啉;和步骤(2):在溶剂中在酸、亚硝酸钠和催化量的催化剂存在的情况下,使具有4-羟基-6-氨基喹唑啉与呋喃甲醛反应,制得所述中间体。优选的是,在步骤(1)中,所述溶剂为水和/或乙醇。另外优选的是,反应在80℃至85℃进行,所述催化剂为钯碳。在一些更具体的实施方式中,所述步骤(1)可以通过如下方式进行:向反应容器中加入4-羟基-6-硝基喹唑啉、水和钯碳,搅拌均匀后加入水合肼,升温至80℃回流反应2.5小时,趁热过滤得到滤液,除去滤液中的至少一部分(例如80体积%)溶剂后降温结晶,过滤晾干得到固体形式的所述4-羟基-6-氨基喹唑啉。在另外一些优选的实施方式中,在步骤(2)中,所述催化剂选自由如下催化剂组成的组:CuCl2和/或其水合物;Cu(OCOCH3)2和/或其水合物;Cu2Cl2;硝酸铈铵;钯碳、钯和/或其衍生物,更优选所述催化剂为CuCl2和/或其水合物和Cu2Cl2。本发明对催化剂的用量没有特别的限制,但是优选所述催化剂的用量为0.01至1当量(例如0.02、0.2、0.7或1当量),更优选为0.1到0.7当量。在另外一些优选的实施方式中,呋喃甲醛的用量可以为1当量到过量(例如1、2、3、5、6、7、8、9或10当量),更优选呋喃甲醛的用量为2到5当量。在另外一些优选的实施方式中,所述酸为盐酸和/或硫酸,更优选所述酸为盐酸。本发明对步骤(2)中所使用的溶剂没有特别的限制,一般可以使用适用于重氮化反应用的极性溶剂或其混合物。但是优选所述溶剂为选自由水、丙酮、二甲基甲酰胺和二甲基亚砜组成的组,优选所述溶剂为水和丙酮。在一些更具体的实施方式中,所述步骤(2)可以通过如下方式进行:向反应容器中加入水和4-羟基-6-氨基喹唑啉,再加入盐酸并搅拌溶解,降温至0℃,滴加亚硝酸钠水溶液,在0℃反应10分钟,滴加呋喃甲醛和丙酮并搅拌,再滴加氯化铜水溶液并搅拌,加入钯碳催化剂并在0℃反应2小时,然后升温至室温后反应24小时或直到反应完全,过滤,固体用水洗,用体积比为1:1的甲醇和二氯甲烷的混合溶剂溶解过滤,滤液旋蒸得到产品盐酸盐粗品;母液用乙酸乙酯萃取,水洗乙酸乙酯层,浓缩得到更多固体形式的所述5-(4-羟基喹唑啉)-呋喃-2-甲醛盐酸盐。经用氨水调碱(pH=6-7)乙酸乙酯萃取可得到产品的游离碱。在一些优选的实施方式中,所述方法在步骤(1)之前可以还包括步骤(0):在催化量的催化剂存在的情况下使2-氨基-5-硝基苯甲酸和甲酰胺反应从而制得所述4-羟基-6-硝基喹唑啉。在一些更优选的实施方式中,在步骤(0)中,所述催化剂为硼酸。优选的是,在步骤(0)中,反应在130℃至160℃下进行。在一些更具体的实施方式中,所述步骤(0)可以通过如下方式进行:在反应容器中加入2-氨基-5-硝基苯甲酸,再加入甲酰胺,搅拌升温至110-120℃使固体溶解,再加入硼酸作为催化剂,升温至150℃至160℃反应后反应7小时,降温析出晶体,过滤,从而得到固体形式的所述4-羟基-6-硝基喹唑啉。本发明在第二部分还提供了一种合成拉帕替尼和/或其盐、拉帕替尼中间体和/或所述中间体的药学可接受的盐的方法,所述方法利用5-(4-羟基喹唑啉)-呋喃-2-甲醛进行,并且所述5-(4-羟基喹唑啉)-呋喃-2-甲醛由权利要求1至5中任一项所述的方法合成。在一些优选的实施方式中,所述中间体的盐为盐酸盐、硫酸盐或亚硫酸盐,和/或所述拉帕替尼的盐为二对甲苯磺酸盐。在一些优选的实施方式中,所述中间体选自由为5-(4-氯喹唑啉-6-基)呋喃-2-甲醛和/或5-(4-(3-氯-4-(3-氟苄氧基)苯基氨基)-喹唑啉-6-基)呋喃-2-甲醛。在一些优选的实施方式中,所述方法可以包括如下步骤:步骤(3):使用5-(4-羟基喹唑啉)-呋喃-2-甲醛合成5-(4-氯喹唑啉-6-基)呋喃-2-甲醛盐酸盐;步骤(4):使用5-(4-氯喹唑啉-6-基)呋喃-2-甲醛盐酸盐合成5-(4-(3-氯-4-(3-氟苄氧基)苯基氨基)-喹唑啉-6-基)呋喃-2-甲醛盐酸盐;和/或步骤(5):使用5-(4-(3-氯-4-(3-氟苄氧基)苯基氨基)-喹唑啉-6-基)呋喃-2-甲醛盐酸盐合成拉帕替尼。在一些更为具体的实施方式中,所述步骤(3)可以通过如下方式进行:在氮气保护下使5-(4-羟基喹唑啉)-呋喃-2-甲醛和SOCl2在二甲基甲酰胺存在的情况下反应从而得到5-(4-氯喹唑啉-6-基)呋喃-2-甲醛盐酸盐。在另外一些更为具体的实施方式中,所述步骤(4)可以通过如下方式进行:使5-(4-氯喹唑啉-6-基)呋喃-2-甲醛盐酸盐和3-氯-4-(3-氟-苄氧基)苯胺在乙腈中反应,从而得到5-(4-(3-氯-4-(3-氟苄氧基)苯基氨基)-喹唑啉-6-基)呋喃-2-甲醛盐酸盐;和/或在另外一些更为具体的实施方式中,所述步骤(5)通过如下方式进行:使5-(4-(3-氯-4-(3-氟苄氧基)苯基氨基)-喹唑啉-6-基)呋喃-2-甲醛盐酸盐)和2-(甲磺酰基)乙胺反应从而得到拉帕替尼。在另外一些可选的实施方式中,所述步骤(5)可以进一步包括将所述拉帕替尼与一水合对甲苯磺酸反应从而得到二对甲苯磺酸拉帕替尼。实施例下文将通过实施例的形式对本发明进行进一步的说明,但是这些实施例仅处于描述本发明的目的,而不应理解为对本申请请求保护范围的限制。本发明方法拉帕替尼及其关键中间体的合成路线如下:本发明实施例中所使用的原料和试剂都可以商购获得或者通过已知的合成方法获得。实施例1:4-羟基-6-硝基喹唑啉的合成在反应瓶中加入5g的2-氨基-5-硝基苯甲酸(购自广拓化学(上海)有限公司),再加入约50mL的甲酰胺,搅拌升温至110-120℃后使固体溶解,再加入少量硼酸作为催化剂,升温至150℃至160℃反应,在此温度下反应约7h左右。经过TLC检测(展开剂为二氯甲烷:甲醇=10:1),发现2-氨基-5-硝基苯甲酸反应完全。经自然降温而析出固体(约70℃左右固体析出)。降温至室温后过滤,滤饼水洗一次,得到土黄色固体,将该固体于室温晾干后得到固体4.2g。经TLC显示单点(展开剂甲基叔丁基醚:甲醇/20:1)检测,收率78%,可直接用于下步反应。实施例2:4-羟基-6-氨基喹唑啉的合成依次向250mL反应瓶中加入4g实施例1中合成的4-羟基-6-硝基喹唑啉,60ml的水,60ml的乙醇,0.1g左右的钯碳,搅拌均匀后加入4.8mL的水合肼,升温至80℃左右反应,在此温度下反应约2.5h,经TLC检测(展开剂为二氯甲烷:甲醇=10:1)发现原料和中间态均无剩余后终止反应,趁热过滤,滤饼用50mL80℃左右热水洗涤,合并滤液,蒸馏除去4/5的溶剂降温结晶。降温至10℃左右后过滤,滤饼冷水洗涤一次,常温晾干后获得浅黄色固体约3.3g,经TLC检测显示单点(展开剂甲基叔丁基醚:甲醇/20:1),发现收率97%,可直接用于下步反应。实施例3:5-(4-羟基喹唑啉)-呋喃-2-甲醛的合成向100ml反应瓶中加入水10ml,1g实施例2中合成的4-羟基-6-氨基喹唑啉,再加入1.5ml的浓盐酸,搅拌溶解后获得澄清浅黄透明溶液。冰盐浴下降温至0℃左右后,缓慢滴加0.43g亚硝酸钠于0.3ml的水溶液(0℃冰浴冷处理过),反应液变为红黄色。在冰浴下反应约10min后,缓慢滴加1ml呋喃甲醛和10ml丙酮,滴毕,搅拌2分钟后,缓慢滴加0.7g氯化铜于0.3ml水溶液,反应液逐渐变为土黑色,搅拌2分钟后加入4%(wt/wt)钯碳催化剂(购自日照力德士化工有限公司)。在0℃左右反应约2h后,自然升温至室温后反应约24h或直到反应完全,过滤,滤饼用体积比为1:1的二氯甲烷:甲醇的混合溶剂溶解过滤,干燥(MgSO4)旋蒸得到粗品,母液用乙酸乙酯萃取,萃取完后,乙酸乙酯层水洗两次,浓缩经洗涤的乙酸乙酯层,合并得到粗品黄色固体约1g,经检测,所得5-(4-羟基喹唑啉)-呋喃-2-甲醛的纯度为93%,收率为66%。HNMR(400MHZ,DMSO-d6)δ7.48(d,J=4Hz,1H),7.69(dd,J=4Hz,J=3.4Hz,1H),7.78(d,J=8.6Hz,1H),8.17(d,J=3.4Hz,1H),8.29(dd,J=1.8Hz,J=8.6Hz,1H),8.54(d,J=1.8Hz,1H),9.66(s,1H),12.45(bs,1H).实施例4:5-(4-氯喹唑啉-6-基)呋喃-2-甲醛盐酸盐将100mg实施例3中合成得到的经研磨成细粉状的5-(4-羟基喹唑啉)-呋喃-2-甲醛放置于25ml的单口烧瓶中,加入SOCl2(2ml)和DMF(二甲基甲酰胺)(0.04ml),在氮气保护下,升高温度到80℃,搅拌直到原料反应完毕,减压蒸去SOCl2,加入2ml乙腈,并超声清洗,过滤,滤饼经1ml乙腈清洗,真空干燥后得到104mg黄色固体。产率85%,纯度:90%(HPLC)。实施例5:5-(4-(3-氯-4-(3-氟苄氧基)苯基氨基)-喹唑啉-6-基)呋喃-2-甲醛盐酸盐的合成将50mg原料1(实施例4中合成得到的5-(4-氯喹唑啉-6-基)呋喃-2-甲醛盐酸盐)加入到1.5ml乙腈中,并加入48mg的原料2(如上所示的化合物6,即3-氯-4-(3-氟-苯氧基)苯胺)的乙腈溶液(0.5ml),在70℃搅拌,待原料反应完后,停止加热,冷却到室温(12℃),过滤,滤饼经乙腈(1ml*2)洗涤,真空干燥后得到74.2mg棕色固体。产率86%,纯度:85%(由HPLC测得)。实施例6:拉帕替尼和二对甲苯磺酸拉帕替尼的合成将50mg原料(实施例5中合成得到的5-(4-(3-氯-4-(3-氟苄氧基)苯基氨基)-喹唑啉-6-基)呋喃-2-甲醛盐酸盐)加入到DCM(二氯甲烷)(1ml)中并加入DIEA(N,N-二异丙基乙胺)(25mg,2eq),搅拌使其溶解,并加入2-(甲磺酰基)乙胺(24mg,2eq),于室温下(18℃)搅拌直到原料反应完毕,减压蒸干DCM(二氯甲烷),并加入THF(四氢呋喃)(1ml)使其溶解,加入NaHB(OAc)3(124mg,6eq),室温搅拌直到原料反应完毕,加入Na2CO3水溶液(1ml,5%)并用EA(乙酸乙酯)萃取(1ml*2),合并有机层,用饱和盐水反洗(1ml*1),有机层经干燥,蒸干后得到50mg棕色油状物。将50mg棕色油状物溶于1ml的THF中,滴加一水合对甲苯磺酸(33mg,2eq)的THF溶液(0.5ml),升高温度到50℃,搅拌1小时后,冷却到室温(15℃),抽滤,滤饼用THF洗涤(1ml*2)经干燥后得到49mg黄色固体,将此黄色固体加入到THF/H2O(4/1)1ml中,加热到60℃使其溶解澄清,并缓慢降低温度到室温(15℃)搅拌1小时后,继续降低温度到0℃,搅拌1小时后抽滤,滤饼用THF洗涤(1ml*2),经真空干燥后得到30mg黄色固体。收率:33%,纯度99%(HPLC,1HNMR)。HNMR(400MHZ,DMSO-d6)δ2.28(s,6H),3.13(s,3H),3.45-3.49(t,2H),3.56-3.60(t,2H),4.48(s,2H),5.31(s,2H),6.89(d,J=3.2HZ,1H),7.09(d,J=8HZ,4H),7.21(dt,J=8.8,2HZ,1H),7.26(d,J=3.2HZ,1H),7.35(m,3H),7.47(m,5H),7.60(dd,J=9.2,2.4HZ),7.86(d,J=2.4HZ,1H),7.91(d,J=8.8HZ,1H),8.42(dd,J=8.8,1.6HZ,1H),8.94(s,1H),9.05(s,1H),9.33(br,s,1H),11.41(s,1H)。实施例7根据实施例1所述的方式进行,不同的是反应在135℃进行。经检测,收率为74%。实施例8根据实施例2所述的方式进行,不同的是,反应在85℃进行。经检测,收率为90%。实施例9至12根据实施例3所述的方式进行,不同的是,使用的催化剂及其用量以及所测得的5-(4-羟基喹唑啉)-呋喃-2-甲醛的收率如下:实施例13至16根据实施例3所述的方式进行,不同的是,催化剂用量以及所测得的5-(4-羟基喹唑啉)-呋喃-2-甲醛的收率如下:实施例17至20根据实施例3所述的方式进行,不同的是,呋喃甲醛的用量以及所测得的5-(4-羟基喹唑啉)-呋喃-2-甲醛的收率如下:实施例用量(当量)收率(%)17137%18266%19569%201073%实施例21根据实施例3所述的方式进行,不同的是,所使用的酸为硫酸,并且所测得的5-(4-羟基喹唑啉)-呋喃-2-甲醛的收率为49%。实施例22至24根据实施例3所述的方式进行,不同的是,所使用的溶剂以及所测得的5-(4-羟基喹唑啉)-呋喃-2-甲醛的收率如下。实施例溶剂收率(%)22丙酮66%23二甲基甲酰胺50%24二甲基亚砜46%本发明人还尝试使用如下路线合成:由于产物在酸性条件下可以生成盐,且其水溶性极好,使得用氯化铵和铁粉还原的后处理相对繁琐,而且所得产物容易包含无机盐杂质,难于完全清除。相反,本发明使用的原料2-氨基-5-硝基苯甲酸和甲酰胺价格便宜,在催化剂例如硼酸存在下,加热环合,不涉及其它环合方法中使用的腐蚀性强的浓硫酸;后处理简单,只需过滤洗涤就可以得到纯度较高的产品,可直接用于下步反应;用水合肼还原反应快捷,后处理简单,将大部分溶剂蒸出,降温产品析出。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1