一种用于有机电致发光器件的主体材料的制作方法
本发明属于化学合成技术领域,具体地讲,涉及一种用于溶液法加工制备有机电致发光器件的可交联的双极性的主体材料。
背景技术:
有机发光二极管(Organic Light-emitting Diode,OLED)是一种基于有机材料的电致发光器件。目前制作有机发光二极管的工艺主要包括真空蒸镀法和溶液加工法。在有机全彩显示领域,国内外研发与生产的公司基本采用真空蒸镀和印刷技术制备显示屏,这是当前国际主流的发展技术。小分子的真空蒸镀技术比较成熟,目前已经实现产业化,并有中小尺寸的全彩色显示屏批量推出,应用到智能手表、手机、平板及电视机等电子设备领域。但是,真空蒸镀技术由于设备投资和维护费用高昂、材料浪费严重、产品良率低等因素导致了有机电致发光制作成本居高不下。传统的真空镀膜设备大多采用的是单点蒸镀源技术,材料的利用率仅有5%。近几年发展的新型的线型蒸镀源技术对材料的利用率有了一定的提升,达到了20%左右。与之相比,印刷技术对材料的利用率能达到90%以上,特别是喷墨打印(Ink-jet printing)可以根据需求直接将材料喷涂在基板上,不会造成材料的扩散和浪费,理论上的材料利用率可以达到99%。目前,溶液法制作有机电致发光的工艺主要包括旋涂(Spin-coating)、喷墨打印、卷对卷(Roll to Roll)、凹版印刷(Gravure printing)、丝网印刷(Screen printing)等。旋涂工艺具有工艺简单和设备投资费用低等优点,在实验室获得广泛的使用。印刷工艺具有能够方便地实现图案化、印刷速度快、成形面积大的优点,因此十分适于大规模的工业化生产。
近几年来对双极性主体材料的研究显示该策略是实际可行的,可以有效地提高有机发光二极管器件的性能。将此双极性的特点结合上交联基团可以解决溶液法制备多层结构的难题。
虽然溶液法由于其简单的制作工艺而被广泛地应用于有机电致发光器件的制备中,但是溶液法制备的有机电致发光的器件也存在一些问题。采用溶液法加工时,上面一层的溶液会侵蚀下层有机功能层,造成下层有机功能层的表面粗糙度变大,甚至于直接破坏下层有机层。由于表面形貌上的缺陷引发的功能 层界面的接触问题会对器件的效率及寿命造成严重影响,不利于器件性能的提高。
技术实现要素:
为了解决上述现有技术的不足,本发明提供一种用于有机电致发光器件的主体材料,该主体材料可以用于溶液法加工制备有机电致发光器件,并可以具有可交联和双极性的特点。
根据本发明的示例性实施例,一种用于有机电致发光器件的主体材料包括由式1表示的结构,
R1、R2和R3中的每个均独立地选自于由氢原子、下面的式2表示的基团和下面的式3表示的基团组成的组,
其中,R选自于由氢原子、氰基、三氟甲基、C1-C30烷氧基和C1-C30烷基组成的组,R4为氢原子或交联基团C,所述交联基团C选自于式C-1至式C-5:
n为0至5的整数,式C-1和式C-4中的R与上述定义相同,m为0至5的整数,*指示结合位。
与传统的主体材料相比,据本发明的示例性实施例的用于有机电致发光器件的主体材料使得可以使用印刷的方法形成有机电致发光器件,而且使得后续的功能层也可以通过印刷的方法形成。另外,据本发明的示例性实施例的用于有机电致发光器件的主体材料具有很强的抗溶剂侵蚀的能力。另外,据本发明的示例性实施例的用于有机电致发光器件的主体材料具有双极性的特点,对电子和空穴具有更加平衡的传输性能,因此有利于有机电致发光器件的性能的提升。
附图说明
通过参照附图详细描述示例性实施例,特征对于本领域技术人员来讲将变得明显,在附图中:
图1示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在二氯甲烷淋洗前后的吸收光谱的曲线图;
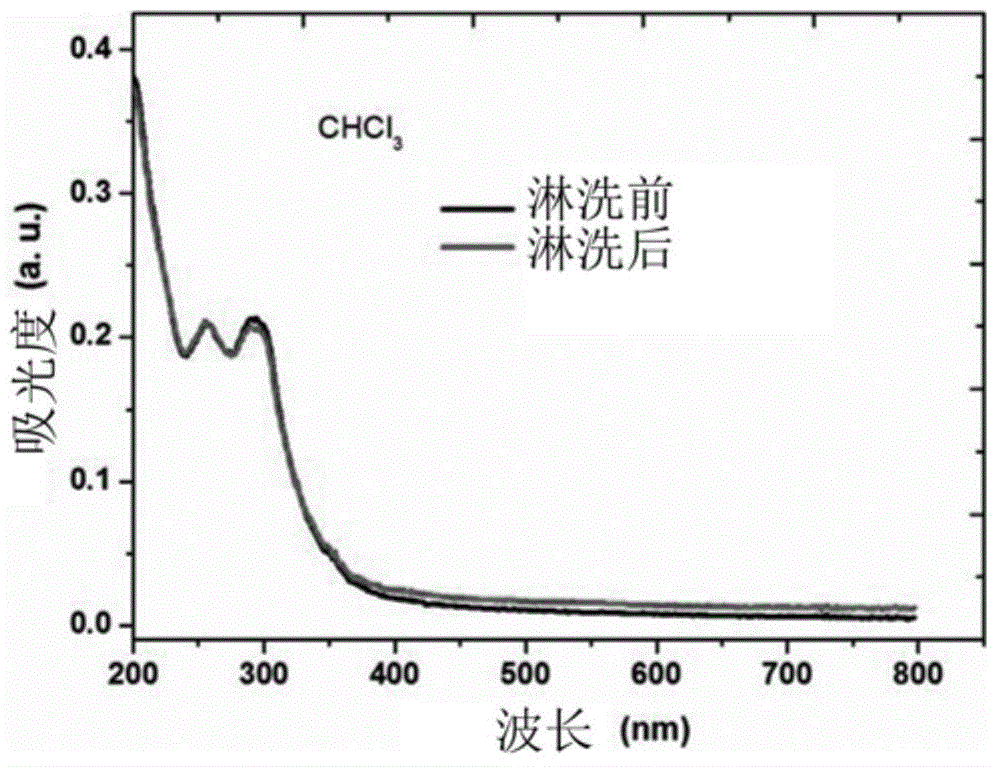
图2示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在氯仿淋洗前后的吸收光谱的曲线图;
图3示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在甲苯淋洗前后的吸收光谱的曲线图;
图4示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在二甲苯淋洗前后的吸收光谱的曲线图;
图5示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在1,2-二氯苯淋洗前后的吸收光谱的曲线图;
图6示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料 在四氢呋喃淋洗前后的吸收光谱的曲线图;
图7示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在交联固化前后的电荷传输性能量表征的结果;
图8示出采用根据本发明的示例性实施例的用于有机电致发光器件的主体材料形成的有机发光二极管器件的电流密度-电压-亮度的曲线图;
图9示出采用根据本发明的示例性实施例的用于有机电致发光器件的主体材料形成的有机发光二极管器件的电压效率的曲线图;
图10示出采用根据本发明的示例性实施例的用于有机电致发光器件的主体材料形成的有机发光二极管器件的电致发光光谱图。
具体实施方式
现在将参照附图在下文中更充分地描述示例实施例;然而,示例实施例可以以不同的形式实施,且不应当被解释为局限于这里阐述的实施例。相反,提供这些实施例使得本公开将是彻底的且完整的,并将向本领域技术人员充分地传达示例性实施方案。
根据本发明的示例性实施例的用于有机电致发光器件的主体材料包括由式1表示的结构,
R1、R2和R3中的每个均独立地选自于由氢原子、下面的式2表示的基团和下面的式3表示的基团组成的组,
其中,R选自于由氢原子、氰基、三氟甲基、C1-C30烷氧基和C1-C30烷基组成的组,R4为氢原子或交联基团C,所述交联基团C选自于式C-1至式C-5:
n为0至5的整数,式C-1和式C-4中的R与上述定义相同,
m为0至5的整数,*指示结合位。
根据示例性实施例,R可以选自于由氢原子、氰基、三氟甲基、C1-C10烷氧基和C1-C10烷基组成的组。可选地,R选自于由甲基、乙基、丙基、异丙基和叔丁基组成的组。
根据示例性实施例,m可以为0或1。
根据示例性实施例,由式1表示的结构可以为式1-1至式1-12中的任意一者:
根据示例性实施例,由式2表示的结构可以为式2-1至式2-6中的任意一者:
*指示结合位。
根据示例性实施例,R1可以为由式3表示的基团,R2可以为氢原子,R3可以为由式2表示的基团,其中,R为叔丁基,R4为交联基团C-2,m=1,n=1。
根据示例性实施例,R1和R2可以均为由式2表示的基团,R3可以为氢原子,其中,R为叔丁基,R4为交联基团C-2,n=1。
根据示例性实施例,R1、R2和R3可以均为由式2表示的基团,其中,R为叔丁基,R4为交联基团C-2,n=1。
根据示例性实施例,所述主体材料可以是下面的化合物1至化合物24中的一个:
根据本发明的示例性实施例的用于有机电致发光器件的主体材料是可印刷的且具有强的抗溶剂侵蚀的能力,并且具有双极性的特点,能够有效地提高对载流子的传输性能。通过对根据本发明的示例性实施例的用于有机电致发光器件的主体材料的表征,证实该材料对有机溶剂具有很好的抗性,并且有机电致发光器件的表征显示该材料具有普通主体材料不具有的优越性。
根据本发明的示例性实施例的用于有机电致发光器件的主体材料具有能够交联固化的特点,可以有效地解决在溶液法制备有机电致发光器件的过程中的上层对下层有机功能层的侵蚀问题。在此,需要特别指出的是,与传统的主体材料不同的是,本发明中的主体材料同时具备有双极性和可交联固化两个特点,双极性能够保证器件具有较好的性能,可交联固化可以使得与发光层相邻的电子传输层也能进行溶液法制作,使得有机电致发光器件的功能层实现了全溶液法制备的特点。
提供下面的示例以突出一个或更多个实施例的特性,但将理解的是,示例不应被解释为限制实施例的范围。此外,将理解的是,实施例不限于在示例中描述的具体细节。
示例
合成示例1:化合物1的合成
中间体I-1的合成
分别称取27.17g(126.34mmol)4-溴苯甲酰肼和4.73g(10.49mmol)碳酸钾倒入500mL的三颈烧瓶中,然后加入240mL NMP,在氮气保护下52℃下搅拌15分钟后,分四次加入25.02g(126.34mmol)4-(氯甲酰基)苯甲酸甲酯,间隔为5分钟/次。在氮气保护下加热搅拌过夜。反应停止冷却至室温后,将反应混合物在搅拌下慢慢倒入800mL水中,待白色固体析出后,进行抽滤,用水淋洗沉淀4次,分三批收集沉淀,在加热板上在90℃下烘干,以获得44.06g中间体I-1(产率92.7%)。13C NMR(101MHz,DMSO)δppm:10.77-10.65(d,2H),8.13-8.00(m,4H),7.90-7.84(dt,2H),7.79-7.72(dt,2H),3.90(s,3H).13C NMR(101MHz,DMSO)δ165.61,165.05,164.90,136.54,132.38,131.62,131.51,129.55,129.33,127.87,125.86,125.74,52.43.
合成中间体I-1的反应方程式:
中间体I-2的合成
量取38.03mL对叔丁基苯胺和110mL邻二氯苯于500mL二颈烧瓶中,在氮气保护下,加入5.10mL三氯化磷,100℃搅拌反应40分钟后,加入15.0g(39.8mmol)二酰肼,升温至150℃搅拌过夜。反应停止冷却至室温后,用二氯甲烷萃取,抽滤。将粗产品拌硅胶柱层析分离,淋洗剂为二氯甲烷/乙酸乙酯(20:1),以获得11.20g中间体I-2(产率57.4%)。13C NMR(101MHz,cdcl3)δppm:7.96-7.91(dt,2H),7.51-7.39(m,6H),7.30-7.26(dt,2H),7.08-7.04(dt,2H),3.89(s,3H).13C NMR(101MHz,cdcl3)δ166.54,154.49,154.32,153.91,132.10,131.86,131.18,131.11,131.06,131.04,130.27,129.72,128.68,128.06,127.33,127.25,125.88,124.54,120.46,52.45,35.13,31.38.
合成中间体I-2的反应方程式:
中间体2-1的合成
分别称取15.25g(40.1mmol)3-溴-9-苯甲酸甲酯咔唑、15.27g(60.2mmol)联硼酸频哪酯和11.81g(120.3mmol)乙酸钾,加入到500mL二颈烧瓶中,然后加入200mL1,4-二氧六环。在氮气保护下,加热至115℃搅拌均匀后加入2.93g(4.01mmol)PdCl2(dppf),在氮气保护下搅拌回流18h。停止反应后静置至室温,用装有硅胶的砂芯漏斗进行真空抽滤后,用二氯甲烷多次淋洗硅胶至无产物,收集滤液进行旋蒸得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂为正己烷/乙酸乙酯(20:1),分离得到13.651g中间体2-1(产率79.8%)。1H NMR(400MHz,DMSO)δppm:8.64(s,1H),8.33-8.26(dt,2H),8.21-8.15(dt,1H)7.90-7.85(dd,1H),7.72-7.63(dt,2H),7.48-7.39(m,3H),7.36-7.29(m,1H),3.99(s,3H),1.41(s,12H).13C NMR(101MHz,DMSO)δ161.55,137.60,137.02,135.63,127.90,126.61,124.09,123.02,121.70,121.40,119.17,118.70,116.11,115.86,104.98,104.32,78.95,78.71,72.61,72.29,71.97,47.56,20.29,20.16,20.04.
合成中间体2-1的反应方程式:
中间体I-3的合成
分别称取4.0g(9.36mmol)对溴三氮唑和4.59g(9.36mmol)咔唑苯硼酸酯并加入到250mL三颈烧瓶中,然后加入120mL甲苯、30mL乙醇和14.04mL浓度为2M的碳酸钾水溶液。在氮气保护下,加热至100℃搅拌均匀后加入1.08g(0.94mmol)催化剂Pd(PPh3)4,在氮气保护下搅拌回流过夜。反应停止冷却至室温后,加入450mL水进行萃取,然后进行分液,有机相再次用水萃取除去无机盐等,水相用乙酸乙酯萃取至无产物后,一并用无水硫酸钠干燥,旋蒸得到粗产物。粗产物用柱层析法分离,淋洗剂为二氯甲烷/乙酸乙酯(10:1),分离后得到4.32g中间体I-3(产率:65.0%)。1H NMR(400MHz,DMSO)δppm:8.36-8.25(m,3H),8.19-8.12(dt,1H),8.00-7.91(dt,2H),7.70-7.61(m,5H),7.59-7.40(m,9H),7.36-7.29(m,1H),7.18-7.13(dt,2H),3.99(s,3H),3.90(s,3H),1.37(s,9H).13C NMR(101MHz,cdcl3)δ166.41,166.30,155.03,153.95,153.60,142.99,141.76,140.73,140.00,133.03,132.63,132.18,132.09,131.99,131.90,131.41,131.10,130.89,130.75,130.73,130.72,130.69,129.55,129.09,128.84,128.52,128.42,127.08,126.31,125.44,124.69,124.37,123.73,120.46,45.05,34.96,31.27,31.23.
合成中间体I-3的反应方程式:
中间体I-4的合成
称取0.91g(24.08mmol)LiAlH4于250mL二颈烧瓶中,加入50mL四氢呋喃,然后在氮气保护下置于0℃下搅拌30分钟,再用恒压漏斗滴加入4.28g(6.02mmol)咔唑-p-三唑甲酸甲酯的四氢呋喃溶液100mL,在氮气保护下在0℃下搅拌2h后,将其转至室温下搅拌,加入无水甲醇淬灭反应至体系无气泡放出,旋转蒸发得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂为纯乙酸乙酯,分离得到3.94g中间体I-4(产率70%)。1H NMR(400MHz,DMSO)δppm:8.65-8.60(d,1H),8.37-8.29(dt,1H),7.81-7.74(m,3H),7.65-7.25(m,20H),5.38(t,1H),5.24(t,1H),4.68-4.62(d,2H),4.51-4.45(d,2H),1.23(s,9H).13C NMR(101MHz,dmso)δ154.47,154.08,152.49,144.15,142.26,141.65,140.73,140.02,135.09,132.40,130.95,128.79,128.20,128.09,127.90,126.63,126.45,126.32,126.26,125.62,125.57,125.56,125.40,125.25,125.11,123.38,122.81,120.19,118.79,115.85,110.04,62.50,62.38,34.62,30.99.
合成中间体I-4的反应方程式:
化合物1的合成
称取0.29g(12.0mmol)NaH于250mL二颈烧瓶中,在氮气保护下加入10mL无水DMF,置于室温中搅拌,然后用恒压漏斗滴加入2.62g(4.0mmol)咔唑-p-三唑甲醇的无水DMF溶液100mL,在氮气保护下在室温下搅拌3h后,将反应体系转至0℃下搅拌15min,然后用注射器滴加入1.83g(12.0mmol)4-氯甲基苯乙烯的无水DMF溶液5mL,低温搅拌30min后,再转至60℃下在氮气保护中加热搅拌反应过夜。反应停止冷却至室温后,用无水甲醇淬灭反应至体系无气泡放出,旋转蒸发出溶剂得到粗产品。1H NMR(400MHz,CDCl3)δppm:8.31(s,1H),8.17-8.11(d,1H),7.68-7.59(m,5H),7.58-7.52(m,4H),7.50-7.28(m,18H),7.08-7.00(d,2H),6.82-6.64(m,2H),5.82-5.71(ddd,2H),5.30-5.21(t,2H),4.68(s,4H),4.58-4.51(d,4H),1.30(s,9H).13C NMR(101MHz,cdcl3)δ154.92,153.41,141.34,140.65,137.83,137.59,137.43,137.14,136.78,136.46,131.84,130.70,130.66,130.64,129.71,129.41,129.24,128.05,127.43,127.29,126.93, 126.84,126.32,126.28,125.21,123.94,123.30,120.23,118.81,113.93,110.13,72.37,72.29,71.57,71.48,34.87,31.16.
合成化合物1的反应方程式:
合成示例2:化合物3的合成
中间体I-5的合成
分别称取9.0g(41.85mmol)3-溴苯甲酰肼和1.45g(10.49mmol)碳酸钾并倒入250mL三颈烧瓶中,然后加入100mL NMP,在氮气保护下在52℃下搅拌15分钟后,分三次加入8.31g(41.85mmol)4-(氯甲酰基)苯甲酸甲酯,间隔为5分钟/次。在氮气保护下加热搅拌过夜。反应停止冷却至室温后,将反应混合物在搅拌下慢慢倒入400mL水中,进行抽滤,沉淀多次用水淋洗,收集沉淀,90℃下烘干得到14.62g中间体I-5(产率93.1%)。13C NMR(101MHz,DMSO)δppm:10.79-10.67(d,2H),8.10-8.07(m,3H),8.05-8.01(m,2H),7.94-7.88(dt,1H),7.84-7.77(ddd,1H),7.50(t,1H),3.88(s,3H).13C NMR(125MHz,DMSO)δ167.35,166.90,166.63,136.25,134.78,133.53,132.61,131.46,130.78,129.97, 128.56,126.90,121.44,52.08.
合成中间体I-5的反应方程式:
中间体I-6的合成
分别量取37.0mL对叔丁基苯胺和100mL邻二氯苯于250mL二颈烧瓶中,在氮气保护下,加入5.0mL三氯化磷,在100℃下搅拌反应40分钟后,加入14.6g(38.7mmol)m-二酰肼,升温至150℃搅拌过夜。反应停止冷却至室温后,用二氯甲烷萃取,过滤,不溶物用二氯甲烷多次淋洗后,将滤液进行旋蒸得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂为二氯甲烷/乙酸乙酯(7:1),得到13.0g中间体I-6(产率68.5%)。1H NMR(400MHz,CDCl3)δppm:7.99-7.95(dt,2H),7.57-7.53(dt,2H),7.51-7.45(m,4H),7.43-7.37(dt,1H),7.20-7.14(m,1H),7.12-7.04(m,2H),3.91(s,3H),1.36(s,9H).13C NMR(101MHz,CDCl3)δ166.53,154.24,154.06,154.02,132.85,132.01,131.64,131.17,131.09,130.06,129.76,128.79,128.70,127.36,127.27,122.53,52.45,35.15,31.37.
合成中间体I-6的反应方程式:
中间体I-7的合成
分别称取7.10g(14.48mmol)间溴三氮唑和6.19g(14.48mmol)咔唑苯硼酸酯于500mL的三颈烧瓶中,然后加入200mL甲苯、50mL乙醇和21.7mL浓度为2M的碳酸钾水溶液。在氮气保护下,加热至105℃搅拌均匀后加入1.68g(1.45mmol)催化剂Pd(PPh3)4,在氮气保护下搅拌回流过夜。反应停止冷却至室温后,进行抽滤,不溶物用二氯甲烷淋洗。滤液用水洗涤,然后进行分液,水相用二氯甲烷萃取至无产物后,用无水硫酸钠干燥,旋蒸得到粗产物。粗产物用柱层析法分离,淋洗剂为二氯甲烷/乙酸乙酯(7:1)。分离后得到6.785g中间体I-7(产率:66.0%)。1H NMR(400MHz,DMSO)δppm:8.19-8.12(m,3H),8.19-8.12(dt,1H),8.02-7.93(dt,2H),7.78(s,1H),7.75-7.65(m,3H),7.58-7.53(dt,2H),7.51-7.40(m,7H),7.36-7.30(m,2H),7.20-7.12(dt,2H),4.00(s,3H),3.91(s, 3H),1.34(s,9H).13C NMR(101MHz,cdcl3)δ166.57,166.48,155.43,154.11,153.71,141.97,140.83,139.96,133.01,132.44,131.53,131.28,131.07,130.84,130.76,129.72,129.10,128.94,128.76,128.68,127.85,127.50,127.23,127.09,126.65,126.40,125.47,124.48,123.90,120.93,120.67,119.02,110.10,52.51,52.43,35.11,31.37.
合成中间体I-7的反应方程式:
中间体I-8的合成
称取0.64g(16.9mmol)LiAlH4于250mL二颈烧瓶中,加入50mL四氢呋喃,然后在氮气保护下置于0℃下搅拌30min后,用恒压漏斗滴加入3.00g(4.2mmol)咔唑-m-三唑甲酸甲酯的四氢呋喃溶液70mL,在氮气保护下在0℃下搅拌2h后,将其转至室温下搅拌,加入无水甲醇淬灭反应至体系无气泡放出,旋转蒸发得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂为纯乙酸乙酯,分离得到2.158g中间体I-8(产率78.5%)。1H NMR(400MHz,DMSO)δppm:8.43(s,1H),8.34-8.26(d,1H),7.85-7.78(dt,1H),7.65-7.25(m,20H),5.38(t,1H),5.24(t,1H),4.68-4.62(d,2H),4.51-4.45(d,2H),1.23(s,9H).13C NMR(101MHz,dmso)δ154.43,154.22,152.50,144.20,142.28,140.71,139.83,135.06,132.49,131.39,129.19,128.25,128.04,128.00,127.79,127.62,126.66,126.57,126.49,126.30,126.25,125.61,125.37,124.79,123.32,122.69,120.78,120.16,118.58,109.90,109.76,62.49,62.39,34.59,30.89.
合成中间体I-8的反应方程式:
化合物3的合成
称取0.220g(9.16mmol)NaH于250mL二颈烧瓶中,在氮气保护下加入10mL无水DMF,置于室温中搅拌,然后用恒压漏斗滴加入2.0g(3.05mmol)咔唑-m-三唑甲醇的无水DMF溶液75mL,在氮气保护下在室温下搅拌3h后,将反应体系转至0℃下搅拌15min,然后用注射器滴加入1.398g(9.16mmol)4-氯甲基苯乙烯的无水DMF溶液5mL,低温搅拌30min后,再转至60℃下在氮气保护中加热搅拌反应过夜。反应停止冷却至室温后,用无水甲醇淬灭反应至体系无气泡放出,旋转蒸发出溶剂得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂为二氯甲烷/乙酸乙酯(1:1),分离得到0.436g化合物3(产率16.1%)。1H NMR(400MHz,CDCl3)δppm:8.27(s,1H),8.19-8.15(d,1H),7.79(s,1H),7.75-7.70(dt,1H),7.65-7.53(q,4H),7.51-7.29(m,23H),7.19-7.14(dt,2H),6.80-6.68(m,2H),5.83-5.72(ddd,2H),5.31-5.23(ddd,2H),4.69(s,4H),4.58-4.52(d,4H),1.33(s,9H).13C NMR(101MHz,CDCl3)δppm:155.01,154.71,153.10,141.92,141.29,140.41,139.78,137.73,137.62,137.49,137.18,137.14,136.87,136.46,132.62,132.36,130.66,130.64,130.61,130.59,129.21,128.82,128.26,128.06,128.04,127.69,127.53,127.46,126.90,126.87,126.81,126.33,126.28,126.21,125.15,123.86,123.35,120.37,120.14,118.73,113.94,113.90,109.93,72.34,72.20,71.60,71.51,34.89,31.23,29.68.
合成化合物3的反应方程式:
合成示例3:化合物9的合成
中间体I-9的合成
分别称取20.0g(40.78mmol)间溴三氮唑、15.532g(61.17mmol)联硼酸频哪酯和12.06g(122.34mmol)乙酸钾于500ml二颈烧瓶中,然后加入300ml 1,4-二氧六环。在氮气保护下,加热至115℃,搅拌均匀后加入2.983g(4.078mmol)PdCl2(dppf),在氮气保护下搅拌回流5h。停止反应后静置至室温后真空抽滤,用DCM淋洗不溶物后,用400ml水洗反应液两次,然后用200ml二氯甲烷萃取水相,将有机相收集,用无水硫酸钠干燥,旋蒸得到粗产物。粗产物用柱层析法分离,淋洗剂为正己烷/乙酸乙酯(1:1),分离后得到19.115g中间体I-9(产率87.2%)。
1HNMR(400MHz,CDCl3)δppm:7.98-7.89(d,3H),7.80-7.74(dd,1H),7.55-7.48(d,2H),7.47-7.43(m,1H),7.43-7.37(m,2H),7.31-7.26(d,1H),7.10-7.01 (m,2H),3.94-3.85(d,3H),1.36-1.30(s,9H),1.30-1.25(s,12H)
13C NMR(101MHz,CDCl3)δppm:166.59,155.56,153.96,153.38,136.00,135.43,132.23,131.52,131.48,130.89,129.65,128.67,127.79,127.35,126.98,126.33,84.02,52.39,45.17,35.00,31.36,24.96。MS(ESI,m/z)[M+H]+calcd for C32H36BN3O4,537.28;found,538.2880.
合成中间体I-9的反应方程式:
中间体I-10的合成
分别称取0.280g(0.70mmol)3-溴-9-(4-溴苯)咔唑和0.623g(1.16mmol)间硼酯三氮唑于100ml的二颈烧瓶中,然后加入24ml甲苯、6ml四氢呋喃和1ml浓度为2M的碳酸钾水溶液。在氮气保护下,加热至105℃搅拌均匀后,加入催化剂0.081g(0.070mmol)Pd(PPh3)4,在氮气保护下搅拌回流过夜。反应停止冷却至室温后,进行抽滤,不溶物用二氯甲烷淋洗。滤液用60ml水洗两次,然后水相用40ml二氯甲烷萃取两次,收集有机相,用无水硫酸钠干燥,旋蒸得到粗产物。粗产物用柱层析法分离,淋洗剂为正己烷/乙酸乙酯(1:1),分离后得到0.374g中间体I-10(产率60.7%)。
1HNMR(400MHz,CDCl3)δppm:8.30-8.25(s,1H),8.20-8.14(d,1H),8.01-7.94(ddd,4H),7.86-7.82(s,1H),7.77-7.67(m,3H),7.60-7.46(m,14H),7.46-7.28(m,7H),7.22-7.13(dd,4H),3.93-3.89(d,6H),1.39-1.35(s,9H),1.35-1.33(s,9H)。13C NMR(101MHz,CDCl3)δppm:166.56,166.54,155.52,155.08,154.17,154.12,153.78,153.60,142.10,141.29,140.42,140.17,139.42,137.12,132.55,132.51,132.45,131.37,131.24,131.06,130.97,129.74,129.69,129.43,129.01,128.72,128.60,128.46,128.32,127.90,127.51,127.47,127.31,127.29,127.25,127.17,126.90,126.43,125.33,124.09,123.55,120.57,120.45,118.94,110.03,52.43,35.14,35.08,32.04,31.42,31.36,29.81,29.77,29.47,25.14,22.80,14.24。MS(ESI,m/z)[M+H]+calcd for C70H59N7O4,1061.46;found,1062.4679.
合成中间体I-10的反应方程式:
中间体I-11的合成
称取2.4M LiAlH49.42ml(22.60mmol)于500ml二颈烧瓶中,加入10ml四氢呋喃,然后在氮气保护下置于0℃下搅拌30min后,用恒压漏斗滴加入5.766g(5.428mmol)3,9-二(3-三唑甲酸甲酯)咔唑的四氢呋喃溶液200ml,在氮气保护下0℃中搅拌2h后,将其转至室温下搅拌,加入无水甲醇淬灭反应至体系无气泡放出,体系为澄清透明状,旋转蒸发得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂为纯二氯甲烷/甲醇(10:1),分离得到纯产品3.992g中间体I-11(产率73.1%)。
1HNMR(400MHz,CDCl3)δppm:8.26-8.24(t,1H),8.18-8.13(dt,1H),7.81-7.77(d,1H),7.74-7.69(dt,1H),7.69-7.67(d,1H),7.67-7.64(d,1H),7.55-7.27(m,24H),7.17-7.15(d,1H),7.15-7.12(q,2H),7.12-7.10(m,1H),4.73-4.69(d,4H),1.36-1.33(s,9H),1.33-1.30(s,9H)。13C NMR(101MHz,CDCl3)δppm:155.06,155.00,154.93,154.63,153.46,153.31,143.22,143.15,142.02,141.29,140.41,140.11,139.44,137.09,132.61,132.58,132.53,130.61,129.40,128.97,128.51,128.46,128.35,128.31,127.88,127.58,127.54,127.36,127.23,127.16,127.03,126.86,126.82,126.42,125.751,125.67,125.35,124.08,123.56,120.44,110.03,64.52,35.10,35.04,31.43,31.37。MS(ESI,m/z)[M+H]+calcd for C68H59N7O2,1005.47;found,1006.481.
合成中间体I-11的反应方程式:
化合物9的合成
称取0.398g(9.95mmol)的60wt%NaH于250ml二颈烧瓶中,在氮气保护下,用注射器滴入2.0g(1.99mmol)的3,9-二(3-三唑甲醇)咔唑的80ml无水DMF溶液,在氮气保护下室温中搅拌3h后,将反应体系转至0℃下搅拌15min后,用注射器滴加入1.518g(9.95mmol)4-氯甲基苯乙烯。转至60℃下在氮气保护中加热搅拌反应过夜。反应停止冷却至室温后,用无水甲醇淬灭反应至体系无气泡放出,体系为澄清透明状后,加入200ml二氯甲烷,用400ml水洗三次分液,收集有机相,用无水硫酸钠干燥,旋转蒸发出溶剂得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂正己烷/乙酸乙酯(1:1),分离得到纯产品1.142g化合物9(产率46.7%)。1H NMR(400MHz,CDCl3)δppm:8.28(d,1H),8.18(d,1H),7.83(d,1H),7.75–7.70(m,2H),7.70–7.66(m,1H),7.58–7.47(m,12H),7.46(t,2H),7.44(dd,2H),7.41(s,2H),7.40–7.37(m,6H),7.32(qd,9H),7.21–7.19(m,1H),7.17(q,2H),7.16–7.14(m,1H),6.72(dd,2H),5.75(dt,2H),5.25(dt,2H),4.55(dt,8H),1.37(d,9H),1.34(d,9H)。13C NMR(101MHz,CDCl3)δppm:155.06,154.84,154.79,154.62,153.34,153.17,141.91,141.20,140.32,140.01,139.99,139.91,139.37,137.54,137.52,137.16,137.00,136.49,132.64,132.59,132.52,132.15,132.05,129.67,129.63,129.32,128.86,128.62,128.50,128.37,128.22,128.09,127.77,127.64,127.60,127.54,127.51,127.45,127.13,127.09,126.95,126.86,126.33,126.17,125.26,124.01,123.49,120.50,120.36,118.84,113.96,109.95,72.30,72.25,71.54,44.93,35.02,34.95,31.36,31.29,29.73,22.05,14.25,14.19。MS(ESI,m/z)[M+H]+calcd for C86H75N7O2,1237.60;found,1238.6157.
合成化合物9的反应方程式:
合成示例4:化合物21的合成
中间体I-12的合成
分别称取0.576g(1.20mmol)3,6-二溴-9-(4-溴苯)咔唑和2.27g(4.22mmol)间硼酯三氮唑于100ml的二颈烧瓶中,然后加入28ml甲苯、28ml四氢呋喃和7ml浓度为2M的碳酸钾水溶液。在氮气保护下,加热至105℃搅拌均匀后,加 入0.097g(0.084mmol)催化剂Pd(PPh3)4,在氮气保护下搅拌回流过夜。反应停止冷却至室温后,进行抽滤,不溶物用二氯甲烷淋洗。滤液用50ml水洗两次,然后水相用50ml二氯甲烷萃取两次,收集有机相,用无水硫酸钠干燥,旋蒸得到粗产物。粗产物用柱层析法分离,淋洗剂为乙酸乙酯。分离后得到1.177g中间体I-12(产率66.6%)。1H NMR(400MHz,cdcl3)δppm:8.37-8.33(s,2H),8.00–7.98(t,1H),7.98-7.96(d,3H),7.96–7.94(t,2H),7.91-7.88(t,2H),7.77–7.75(m,1H),7.75–7.72(m,1H),7.72–7.64(m,2H),7.62-7.59(t,1H),7.59-7.57(t,1H),7.57–7.51(m,12H),7.50–7.48(d,2H),7.48–7.46(d,3H)7.48-7.38(m,5H),7.33–7.31(m,1H),7.31-7.30(m,1H),7.22–7.13(m,6H),3.91-3.89(d,9H),1.37(s,9H),1.32(s,18H).13C NMR(101MHz,CDCl3)δppm:166.55,166.52,155.50,155.09,154.19,154.11,153.78,153.58,141.96,140.85,140.14,139.53,136.98,132.83,132.52,132.46,131.39,131.25,131.06,130.95,129.73,129.68,129.42,129.00,128.69,128.51,128.44,127.95,127.58,127.51,127.46,127.38,127.30,127.16,127.09,126.88,125.58,124.15,119.10,110.23,52.40,35.15,35.05,31.43,31.35。MS(ESI,m/z)[M+H]+calcd for C96H82N10O6,1470.64;found,1471.6477.
合成中间体I-12的反应方程式:
中间体I-13的合成
称取9.7ml(23.22mmol)2.4M LiAlH4于250ml二颈烧瓶中,在氮气保护下置于0℃下搅拌,用恒压漏斗滴加入5.7g(3.87mmol)3,6,9-三(3-三唑甲酸甲酯)咔唑的四氢呋喃溶液160ml,在氮气保护下0℃中搅拌过夜后,将其转至室温下搅拌,加入无水甲醇淬灭反应至体系无气泡放出,体系为澄清透明状,旋转蒸发得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂为纯二氯甲烷/甲醇(7:1),分离得到纯产品3.879g中间体I-13(产率72.2%)。1H NMR(400MHz,cdcl3)δppm:8.65-8.59(s,2H)7.90-7.80(m,3H),7.79-7.74(m,1H),7.68-7.56(m,9H),7.56-7.48(m,8H),7.48-7.27(m,23H),5.29-5.24(td,3H),4.53-4.46(m,6H),1.31 -1.27(s,9H),1.24-1.20(S,18H).13C NMR(101MHz,CDCl3)δppm:154.43,154.26,152.60,152.47,144.26,144.22,140.62,140.05,138.94,138.58,136.12,132.51,132.46,131.90,128.30,128.25,128.01,127.68,126.75,126.64,126.34,126.31,125.36,123.63,62.38,34.67,34.57,30.97,30.88。MS(ESI,m/z)[M+H]+calcd for C93H82N10O3,1386.66;found,1387.6658.
合成中间体I-13的反应方程式:
化合物21的合成
称取0.130g(3.24mmol)的60wt%NaH于100ml二颈烧瓶中,在氮气保护下,用恒压漏斗滴加入0.60g(0.432mmol)的3,6,9-三(3-三唑甲醇)咔唑的无水DMF溶液25ml,在氮气保护下室温中搅拌3h后,将反应体系转至0℃下搅拌15min,然后用注射器滴加入0.494g(3.24mmol)4-氯甲基苯乙烯。转至60℃下在氮气保护中加热搅拌反应过夜。反应停止冷却至室温后,用无水甲醇淬灭反应至体系无气泡放出,体系为澄清透明状后,加入25ml二氯甲烷,用80ml水洗三次分液,收集有机相,用无水硫酸钠干燥,旋转蒸发出溶剂得到粗产品。将粗产品拌硅胶柱层析分离,淋洗剂正己烷/乙酸乙酯(1:1),分离得到纯产品0.098g化合物21(产率13.1%)。1H NMR(400MHz,cdcl3)δppm:8.38-8.33(s,2H),7.91-7.85(d,2H),7.79-7.72(dd,2H),7.72-7.67(dd,2H),7.60–7.43(m,18H),7.43-7.36(td,12H),7.36–7.28(m,14H),7.23–7.12(m,7H),6.79-6.63(dd,3H),5.80-5.71(d,3H),5.28-5.20(d,3H),4.57-4.52(dd,11H),1.40-1.36(s,9H),1.34-1.29(s,18H).13C NMR(101MHz,CDCl3)δppm:141.93,140.86,139.96,137.64,137.29,136.60,132.92,132.74,128.98,127.75,127.71,127.59,127.19,127.04,126.44,124.17,114.07,77.48,77.36,77.16,76.84,72.37,71.66,35.05,31.48,31.39。MS(ESI,m/z)[M+H]+calcd for C120H106N10O3,1734.84;found,1735.8571.
合成化合物21的反应方程式:
可以基于与以上描述的方法类似的方法来合成化合物5、7、16、19和23。
在下文中,将参照附图和评价示例详细说明根据本发明的示例性实施例的用于有机电致发光器件的主体材料的侵蚀性特性、电荷传输性能。
评价示例1:化合物3的抗溶剂侵蚀性表征
图1示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在二氯甲烷淋洗前后的吸收光谱的曲线图。图2示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在氯仿淋洗前后的吸收光谱的曲线图。图3示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在甲苯淋洗前后的吸收光谱的曲线图。图4示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在二甲苯淋洗前后的吸收光谱的曲线图。图5示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在1,2-二氯苯淋洗前后的吸收光谱的曲线图。图6示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在四氢呋喃淋洗前后的吸收光谱的曲线图。
对化合物3进行交联固化,其条件为:90度,30min;200度,90min。分别研究了固化后膜对如下溶剂的抗侵蚀性能:二氯甲烷(CH2Cl2)、氯仿(CHCl3)、甲苯(Toluene)、二甲苯(Xylene)、1,2-二氯苯(1,2-Dichlorobenzene)和四氢呋喃(THF),通过化合物3的在石英基底上的吸收强度在上述有机溶剂淋洗前后的变化来表征根据本发明的示例性实施例的用于有机电致发光器件的主体材料的抗溶剂侵蚀能力,其结果示出在图1至图6中。
如图1至图6所示,根据本发明的示例性实施例的用于有机电致发光器件的主体材料在交联固化后对这些常用的有机溶剂均具有较好的抗侵蚀性能。
评价示例2:化合物3的电荷传输性能研究
图7示出根据本发明的示例性实施例的用于有机电致发光器件的主体材料在交联固化前后的电荷传输性能量表征的结果。
对于交联对化合物3的电荷传输性能的影响进行了实验研究,器件结构为ITO/PEDOT:PSS/化合物3/TPBI/Liq/Al。
如图7所示,交联对根据本发明的示例性实施例的用于有机电致发光器件的主体材料的电荷传输性能基本没有影响,甚至在高电压下交联后的电荷传输能力有所提高。
类似地,对化合物5、7、16、19和23的侵蚀性特性、电荷传输性能进行测试,发现它们在交联固化后对常用有机溶剂均具有较好的抗侵蚀性能,并且交联对其电荷传输性能基本没有影响。
评价示例3:有机发光二极管器件
图8示出采用根据本发明的示例性实施例的用于有机电致发光器件的主体材料形成的有机发光二极管器件的电流密度-电压-亮度的曲线图。图9示出采用根据本发明的示例性实施例的用于有机电致发光器件的主体材料形成的有机发光二极管器件的电压效率的曲线图。图10示出采用根据本发明的示例性实施例的用于有机电致发光器件的主体材料形成的有机发光二极管器件的电致发光光谱图。
使用化合物3按照如下方法制作有机发光二极管器件:采用旋涂的方式形成阳极修饰层PEDOT:PSS和发光层(EML);在5×10-4Pa的真空条件下采用真空蒸镀方法形成电子传输层和电极。将ITO玻璃超声清洗,然后在紫外-臭氧环境下处理大约15分钟,然后将其装入腔体中。形成的有机发光二极管器件的结构为:ITO/PEDOT:PSS/EML 10%wt(80nm)/TPBI(30nm)/Liq(2nm)/Al。采用KEITHLEY 2400系统测试电源,并采用PR655光谱仪进行测试。测试在大气环境下进行,未作封装处理,测试的结果示出在图8至图10中。如图10所示,该光谱与客体发光材料的光谱是一致的,由此可见交联主体材料不影响客体材料的发光。
通过总结和回顾,根据本发明的示例性实施例的用于有机电致发光器件的主体材料是可印刷的且具有强的抗溶剂侵蚀的能力,并且具有双极性的特点,能够有效地提高对载流子的传输性能。通过对根据本发明的示例性实施例的用于有机电致发光器件的主体材料的表征,证实该材料对有机溶剂具有很好的抗 侵蚀性,并且有机电致发光器件的表征显示该材料具有普通主体材料不具有的优越性。
上面虽然结合实施例对本发明的实施方式做了详细的说明,但是本发明并不限于上述实施方式,本发明所属技术领域的技术人员能够理解,在不脱离本发明宗旨的前提下,在权利要求保护范围内,还可以对上述实施例进行变更或改变等。
- 还没有人留言评论。精彩留言会获得点赞!