利用化学重结晶和物理分离相结合提纯4‑氰基吡啶的方法与流程
本发明涉及一种4-氰基吡啶的提纯方法。
背景技术:
4-氰基吡啶是一种重要的精细化工中间体,用于制备异烟酸、异烟肼等医药农药中间体。4-甲基吡啶是制备4-氰基吡啶的唯一原料,但是纯度>97%的4-甲基吡啶价格较高,所以生产上多使用含有10%左右的3-甲基吡啶的4-甲基吡啶为原料经甲胺化反应制备4-氰基吡啶。但是3-甲基吡啶和4-甲基吡啶的沸点仅相差0.7℃,很难分离,由此得到的是4-氰基吡啶和3-氰基吡啶的混合物。3-氰基吡啶和4-氰基吡啶均是易升华物质,其沸点相差5℃,在减压精馏时直接造成管道堵塞。
技术实现要素:
本发明是要解决3-氰基吡啶和4-氰基吡啶沸点相近,难于分离,在减压精馏时易升华直接造成管道堵塞的问题,提供利用化学重结晶和物理分离相结合提纯4-氰基吡啶的方法。
本发明利用化学重结晶和物理分离相结合提纯4-氰基吡啶的方法,具体步骤如下:
步骤一:化学重结晶:将4-氰基吡啶粗品溶于石油醚与正丁醇的混合溶剂中,冷却后,有固体析出,将固体过滤洗涤干燥后,得到纯度>99%的4-氰基吡啶;再将液体部分减压回收溶剂,得剩余物;
步骤二:物理分离:将步骤一的剩余物加热至50~70℃,体系透明后,自然冷却至30~45℃,保温1~3小时,有固体析出,过滤得固体;该固体为纯度是78%~85%的4-氰基吡啶;
步骤三:将步骤二得到的固体重复步骤一的操作一次,得纯度>99%的4-氰基吡啶;
最后收集步骤一和步骤三得到的纯度>99%的4-氰基吡啶,即为提纯后的4-氰基吡啶。
经过本发明方法4-氰基吡啶的总提出率为92%~99%。
进一步的,步骤一中待提纯的4-氰基吡啶粗品是4-氰基吡啶和3-氰基吡啶的混合物,其中4-氰基吡啶的质量百分含量是75%~92%。
进一步的,步骤一中4-氰基吡啶与混合溶剂的比为1g:1.0mL~2.8mL。
进一步的,步骤一中混合溶剂中石油醚与正丁醇的体积比为(5~10):1。
进一步的,步骤一中冷却温度为0℃~10℃的、冷却时间为1~6h。
进一步的,步骤一中溶剂回收后,剩余物为4-氰基吡啶和3-氰基吡啶的混合物,其中4-氰基吡啶的含量为50%~65%。
本发明的有益效果:
本发明利用石油醚-正丁醇重结晶后所得的晶体中含有95%左右的4-氰基吡啶、5%左右的3-氰基吡啶,为白色固体;用石油醚-正丁醇淋洗固体后,得到纯度是99%~99.5%的4-氰基吡啶。步骤二是将除去溶剂后的4-氰基吡啶和3-氰基吡啶的混合物(含50%~65%的4-氰基吡啶,其余物质是35%~50%的3-氰基吡啶),利用4-氰基吡啶与3-氰基吡啶的凝固点不同,在30℃下,可直接分离出纯度是78%~85%的4-氰基吡啶,可再经过石油醚-正丁醇重结晶后,得到纯度在99%以上的合格品。
本发明方法简单,易操作,对于条件要求较低,能够将3-甲基吡啶和4-甲基吡啶分离,并且4-氰基吡啶的总提出率达到92%~99%,适用于工业化生产,有效解决我国没有此项技术的现状。
附图说明
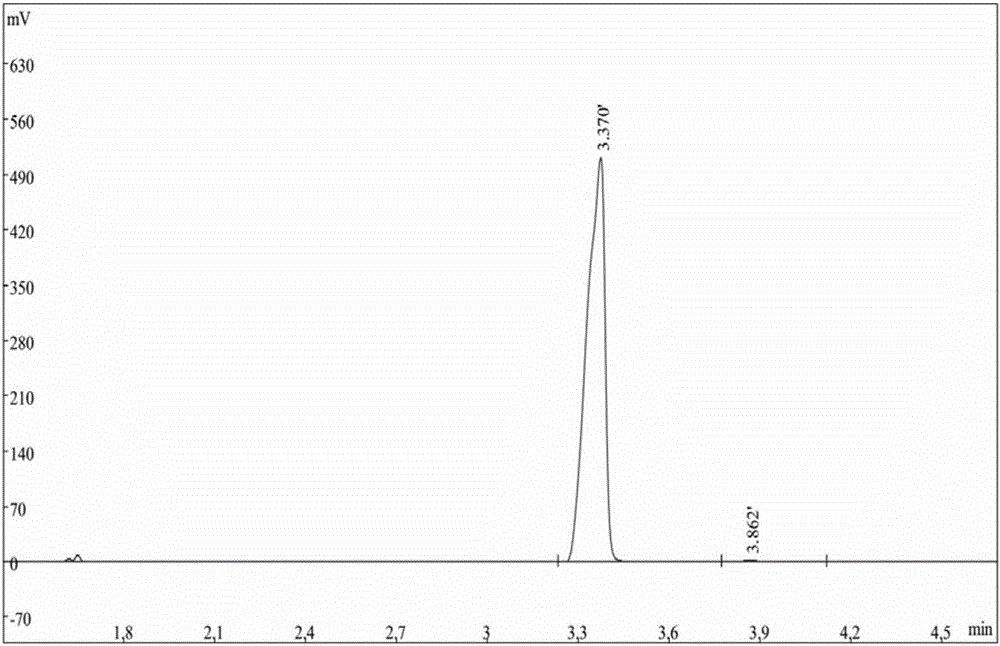
图1为实施例1中4-氰基吡啶待精制粗品的气相色谱图;
图2为实施例1中步骤一提纯后的4-氰基吡啶的气相色谱图;
图3为实施例1中步骤二中82.4%的4-氰基吡啶的气相色谱图;
图4为实施例1中步骤三中提纯后的4-氰基吡啶的气相色谱图;
图5为实施例1中4-氰基吡啶的红外光谱图;
图6为实施例1中4-氰基吡啶的核磁共振氢谱图。
具体实施方式
本发明技术方案不局限于以下所列举具体实施方式,还包括各具体实施方式间的任意组合。
具体实施方式一:本实施方式利用化学重结晶和物理分离相结合提纯4-氰基吡啶的方法,具体步骤如下:
步骤一:化学重结晶:将4-氰基吡啶粗品溶于石油醚与正丁醇的混合溶剂中,冷却后,有固体析出,将固体过滤洗涤干燥后,得到纯度>99%的4-氰基吡啶;再将液体部分减压回收溶剂,得剩余物;
步骤二:物理分离:将步骤一的剩余物加热至50~70℃,体系透明后,自然冷却至30~45℃,保温1~3小时,有固体析出,过滤得固体;
步骤三:将步骤二得到的固体重复步骤一的操作一次,得纯度>99%的4-氰基吡啶;
最后收集步骤一和步骤三得到的纯度>99%的4-氰基吡啶,即为提纯后的4-氰基吡啶。
具体实施方式二:本实施方式与具体实施方式一不同的是:步骤一中待提纯的4-氰基吡啶粗品是4-氰基吡啶和3-氰基吡啶的混合物,其中4-氰基吡啶的质量百分含量是75%~92%。其它与具体实施方式一相同。
具体实施方式三:本实施方式与具体实施方式一不同的是:步骤一中待提纯的4-氰基吡啶粗品是4-氰基吡啶和3-氰基吡啶的混合物,其中4-氰基吡啶的质量百分含量是80%~90%。其它与具体实施方式一相同。
具体实施方式四:本实施方式与具体实施方式一至三之一不同的是:步骤一中待提纯的4-氰基吡啶粗品是4-氰基吡啶和3-氰基吡啶的混合物,其中4-氰基吡啶的质量百分含量是87.1%。其它与具体实施方式一至三之一相同。
具体实施方式五:本实施方式与具体实施方式一至四之一不同的是:步骤一中4-氰基吡啶与混合溶剂的比为1g:1.0mL~2.8mL。其它与具体实施方式一至四之一相同。
具体实施方式六:本实施方式与具体实施方式一至四之一不同的是:步骤一中4-氰基吡啶与混合溶剂的比为1g:1.5mL~2.5mL。其它与具体实施方式一至四之一相同。
具体实施方式七:本实施方式与具体实施方式一至四之一不同的是:步骤一中4-氰基吡啶与混合溶剂的比为1g:2.0mL。其它与具体实施方式一至四之一相同。
具体实施方式八:本实施方式与具体实施方式一至七之一不同的是:步骤一中混合溶剂中石油醚与正丁醇的体积比为(5~10):1。其它与具体实施方式一至七之一相同。
具体实施方式九:本实施方式与具体实施方式一至七之一不同的是:步骤一中混合溶剂中石油醚与正丁醇的体积比为(7~8):1。其它与具体实施方式一至七之一相同。
具体实施方式十:本实施方式与具体实施方式一至九之一不同的是:步骤一中冷却温度为0℃~10℃,冷却时间为1~6h。其它与具体实施方式一至九之一相同。
具体实施方式十一:本实施方式与具体实施方式一至九之一不同的是:步骤一中冷却温度为2℃~8℃,冷却时间为2~5h。其它与具体实施方式一至九之一相同。
具体实施方式十二:本实施方式与具体实施方式一至十一之一不同的是:步骤一中溶剂回收后,剩余物为4-氰基吡啶和3-氰基吡啶的混合物,其中4-氰基吡啶的质量百分含量为50%~65%。其它与具体实施方式一至十一之一相同。
具体实施方式十三:本实施方式与具体实施方式一至十一之一不同的是:步骤一中溶剂回收后,剩余物为4-氰基吡啶和3-氰基吡啶的混合物,其中4-氰基吡啶的质量百分含量为55%~60%。其它与具体实施方式一至十一之一相同。
下面对本发明的实施例做详细说明,以下实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方案和具体的操作过程,但本发明的保护范围不限于下述的实施例。
实施例1:
本实施例利用化学重结晶和物理分离相结合提纯4-氰基吡啶的方法,按以下步骤进行:
步骤一、化学重结晶:将100g 4-氰基吡啶粗品溶于250mL石油醚与正丁醇的混合溶剂中,冷却至10℃后,有固体析出,保温1小时,将固体减压过滤,用上述混合溶剂洗涤干燥后,得到纯度是99.6%的4-氰基吡啶72.3g;再将液体部分减压回收溶剂,得剩余物,剩余物为含量是54.5%的4-氰基吡啶27.7g。
其中4-氰基吡啶粗品是4-氰基吡啶和3-氰基吡啶的混合物,其中4-氰基吡啶的含量是87.1%。石油醚与正丁醇的体积比为10:1。
步骤二:物理分离过程:将步骤一得到的27.7g含量为54.5%的4-氰基吡啶加热至60℃,再自然冷却至30℃,保温1小时,有固体析出,将固体滤出,得到纯度是82.4%的4-氰基吡啶15.2g。
步骤三:将步骤二得到的15.2g纯度是82.4%的4-氰基吡啶溶解于32mL石油醚与正丁醇的混合溶剂中,其中石油醚与正丁醇的体积比为10:1,冷却至10℃后,有固体析出,保温1小时,将固体减压过滤,用上述混合溶剂洗涤干燥后,得到纯度是99.2%的4-氰基吡啶11.6g。最后收集步骤一和步骤三得到的4-氰基吡啶,即为提纯后的4-氰基吡啶。
经过本方法100g 87.1%4-氰基吡啶的总提出量是83.9g,总提出率96.3%。
其中4-氰基吡啶待精制粗品的气相色谱图如图1所示,其中4-氰基吡啶的保留时间3.37min,含量87.1%,3-氰基吡啶的保留时间3.86min,含量12.9%。
步骤一提纯后的4-氰基吡啶的气相色谱图如图2所示;其中4-氰基吡啶的保留时间3.37min,含量99.58%,3-氰基吡啶的保留时间3.86min,含量0.42%。
步骤二中82.4%的4-氰基吡啶的气相色谱图如图3所示;其中4-氰基吡啶的保留时间3.37min,含量82.35%,3-氰基吡啶的保留时间3.86min,含量17.65%。
步骤三中提纯后的4-氰基吡啶的气相色谱图如图4所示;其中4-氰基吡啶的保留时间3.37min,含量99.19%,3-氰基吡啶的保留时间3.86min,含量0.81%。
本发明提纯后的4-氰基吡啶的红外光谱图如图5所示,KBr涂膜法,其中2242cm-1是-CN伸缩振动峰;1593、1545、1497、1416cm-1是吡啶环骨架振动峰;828cm-1是氰基对位取代振动峰。
4-氰基吡啶的核磁共振氢谱图(300MHZ,CDCl3,单位ppm)如图6所示:8.83(d,2H,J=6Hz);7.75(d,2H,J=6Hz)。
实施例2:本实施例所有实验条件和处理方法与实施例1相同,只是混合溶剂的比例改为5:1。产品的总提出率为94.1%,气相色谱检测结果4-氰基吡啶的含量99.5%。
实施例3:本实施例所有实验条件和处理方法与实施例1相同,只是混合溶剂的比例改为7:1。产品的总提出率为95.2%,气相色谱检测结果4-氰基吡啶的含量99.3%。
实施例4:本实施例所有实验条件和处理方法与实施例1相同,只是步骤一和步骤三中冷却的温度改为0℃。产品的总提出率为95.4%,气相色谱检测结果4-氰基吡啶的含量99.3%。
实施例5:本实施例所有实验条件和处理方法与实施例1相同,只是将4-氰基吡啶粗品的含量改为82%。产品的总提出率为93.5%,气相色谱检测结果4-氰基吡啶的含量99.5%。
实施例6:本实施例所有实验条件和处理方法与实施例1相同,只是将4-氰基吡啶粗品的含量改为78%。产品的总提出率为92.2%,气相色谱检测结果4-氰基吡啶的含量99.4%。
实施例7:本实施例所有实验条件和处理方法与实施例1相同,只是将4-氰基吡啶粗品的投料量量改为500g。产品的总提出率为96.8%,气相色谱检测结果4-氰基吡啶的含量99.6%。
实施例8:本实施例所有实验条件和处理方法与实施例1相同,只是将步骤二中冷却的温度改为40℃,步骤二中析出的固体含量为88。2%,此时产品的总提出率为92.0%,气相色谱检测结果4-氰基吡啶的含量99.4%。
- 还没有人留言评论。精彩留言会获得点赞!