一种零背景的平末端克隆载体pRI857及其构建方法和应用与流程
本发明涉及一种零背景的平末端克隆载体pri857及其构建方法和应用,具体涉及一种用温敏型启动子ci857-pr控制抗性基因kanr的表达以此作为筛选标记的pri857克隆载体及其在基因克隆和基因文库构建中的应用,属于生物技术领域。
技术背景
随着现代生物技术的快速发展,通过pcr技术扩增目的dna片段,利用体外重组的方式,将dna序列插入到克隆载体中,实现目的基因的异源表达,在医学,工业和农业生产上取得了重要的成就,也给生物技术的发展开辟了一条新的道路。同时,由于生物信息学的突飞猛进,二代测序技术的问世,这样,阳性克隆的筛选显得尤为重要,而有效的筛选手段是对dna序列测定和后续基因功能验证的必须手段。利用lacz作为筛选标记的蓝白斑筛选系统,通过产生β-半乳糖苷酶将x-gal分解为半乳糖和深蓝色的物质5-溴-4-靛蓝,有色物质可以使细菌呈蓝色(langley,k.e.;etal.(1975)."molecularbasisofbeta-galactosidasealpha-complementation".proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica.72(4):1254–1257.);而阳性克隆则由于外源基因的插入,破坏了lacz的阅读框,使得β-半乳糖苷酶无法正确表达,不能产生蓝色的5-溴-4-靛蓝,故菌落呈白色,通过颜色的差异可以轻易的挑选出正确克隆。然而这种方法有时候会因为有些空载体也显白色,故而也存在限制。
温敏型启动子pr、pl启动子来至于λ噬菌体,受ci857基因的严格调控,30℃下阻遏pr启动子转录,随着温度的提高,阻遏蛋白ci857与pr启动子的结合逐渐减弱,pr启动子的转录则不断提高(villaverdea,etal.fineregulationofci857-controlledgeneexpressionincontinuouscultureofrecombinantescherichiacolibytemperature[j].appliedandenvironmentalmicrobiology,1993,59(10):3485-3487.)。当温度达到37℃时,阻遏蛋白ci857对pr启动子的抑制能力减弱,使得pr启动子能驱动抗性基因的表达,通过这一技术来进行分子克隆,可以获得100%的阳性克隆。
技术实现要素:
针对基因克隆中的实际问题和需求,本发明提供了一种零背景的平末端克隆载体pri857,该零背景平末端快速克隆载体是利用阻遏子ci857和温敏型启动子pr来控制抗生素基因kanr的表达以此作为筛选标记的pri857克隆载体,可以高效克隆平末端pcr产物和任何具有平末端的dna片段。
本发明的技术方案如下:
一种重组质粒pbbr1mcs-pr,为利用pcr扩增质粒pcp20中的pr启动子基因替换pbbr1mcs-2质粒中的kanr的启动子序列,形成全长序列为seqidno.15的重组质粒。
一种重组质粒puc-ci857,为利用pcr扩增质粒pcp20中的ci857基因替换puc19中的lacza基因,形成全长序列为seqidno.16的重组质粒。
一种零背景的平末端克隆载体pri857,为利用pcr扩增重组质粒puc-ci857中的plac-ci857基因和重组质粒pbbr1mcs-pr中的pr-kanr基因替换质粒pkv6中的ampr和ermb-erma基因,pr-kanr基因,plac-ci857基因与质粒pkv6中的dbl-cole1基因顺序连接形成全长序列为seqidno.17的重组质粒,所述的ci857开放阅读框上设计有bfrbi平末端酶切位点。
进一步地,本发明还提供上述零背景的平末端克隆载体pri857的构建方法,包括引物设计、基因克隆和载体构建,利用pcr扩增质粒pcp20中的pr启动子序列来替换质粒pbbr1mcs-2中kanr基因的启动子序列,形成重组质粒pbbr1mcs-pr;利用pcr扩增质粒pcp20中的ci857基因替换质粒puc19中的lazza基因,形成重组质粒puc-ci857;通过合成引物pcr扩增质粒pbbr1mcs-pr中的pr-kanr目的片段,质粒puc-ci857中的plac-ci857目的片段和质粒pkv6中的dbl-cole1目的片段,最后将pr-kanr、plac-ci857、dbl-cole1片段进行顺序连接,得到零背景克隆载体pri857。
本发明利用温敏性启动子元件ci857-pr控制抗性基因kanr的表达,并在ci857开放阅读框上设计bfrbi酶切位点,以用于克隆任意dna片段。在37℃的卡那霉素培养基中,当目的dna片段插入ci857的bfrbi酶切位点后会破坏原有阻遏基因ci857的阅读框,使得pr启动子的抑制被解除,从而kanr基因得以顺利表达,由此可使转化子在30℃的卡那霉素平板上正常生长,而自连产生的空载体,其转化子在30℃培养时会由于kanr基因依然被ci857蛋白抑制,而不能存活在卡那霉素的平板上,导致细胞死亡,从而实现“零背景”的克隆效果。本发明的零背景平末端克隆载体pri857可用于克隆任意pcr产物和平末端dna序列,并且以kanr基因作为筛选标记时克隆阳性率可以达到100%。此外,该克隆载体也可用于基因组文库的高效构建,同时能有效避免空载体干扰,是后基因组时代高通量基因筛选与功能验证的一种有效工具。
附图说明
图1为pbbr1mcs-pr载体的酶切验证图,m表示marker,a表示pbbr1mcs-pr载体经hindiii酶切的条带,b表示pbbr1mcs-pr载体经psti酶切后的条带。
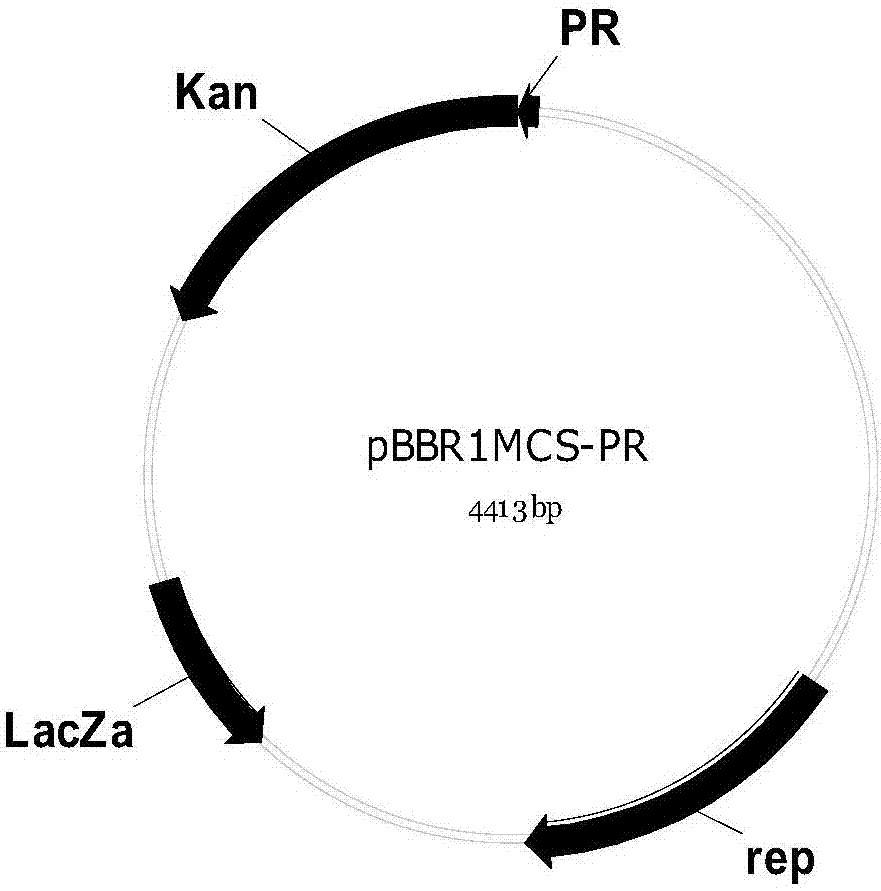
图2为构建完成的pbbr1mcs-pr质粒图谱。
图3为puc-ci857载体的酶切验证图,m表示marker,a表示puc-ci857载体经scai酶切的条带,b表示puc-ci857载体经scai和pcii酶切后的条带。
图4为构建完成的puc-ci857质粒图谱。
图5为pri857载体的酶切验证图,m表示marker,a表示pri857载体经kpni酶切后的条带,b表示pri857载体经psti酶切后的条带。
图6为构建完成的pri857质粒图谱。
图7为dh5α/pri857分别在37℃(a)和30℃(b)培养平板上生长状况。
图8为采用pri857载体用于克隆绿色荧光蛋白基因克隆平板。
图9为利用pri857载体构建基因组文库后经bamhi酶切后的电泳图。
具体实施方式
为了进一步阐明本发明的适用范围,以下集合实施例和附图对本发明加以说明,但该载体的应用不局限本发明所定范围。
本发明所述的质粒pcp20,puc19,pkv6,pbbr1mcs-2,以及大肠杆菌dh5α,dh10β均由商业购买。
实施例一:一种零背景克隆载体pri857的构建方法
1、一种重组质粒pbbr1mcs-pr的构建方法,包括以下步骤:
(1)引物设计:人工设计合成特异性引物序列
prfor:5’-ggttagtatgcagccgtcacagtgctcgttctctggcgtc-3’;
prev:5’-gcatgtactaaggaggttgtatgattgaacaagatggattg-3’
mcsfor:5’-caatccatcttgttcaatcatacaacctccttagtacatgc-3’;
mcsrev:5’-gacgccagagaacgagcactgtgacggctgcatactaacc-3’
(2)pcr扩增:pcr反应体系配制如表1所示。
表1pcr反应体系配制表
pcr程序是98℃预变性30s;98℃变性10s,55℃退火30s,72℃延伸30s,30个循环;最后72℃延伸10min。采用pcp20质粒dna为模板和cifor/cirev引物用以扩增pr启动子目的片段,而采用pbbr1mcs-2质粒dna为模板和mcsfor/mcsrev引物用以扩增rep-lacz-kanr目的片段。所述pr目的片段长度为432bp,rep-lacz-kanr目的片段长度为4061bp。
(3)载体构建:将上述扩增的pr和rep-lacz-kanr目的片段通过琼脂糖凝胶电泳进行分离,并切胶回收。将pr和rep-lacz目的片段用gibsonassembly的方式进行连接,并将连接产物转化至大肠杆菌dh5α中,涂布在含有50μg/ml的卡那霉素lb平板上,于37℃培养18h,挑选克隆于卡那霉素液体培养基中,在30℃培养8h,提取质粒验证,获得质粒pbbr1mcs-pr。质粒pbbr1mcs-pr用hindiii酶切后长度为4413bp的核酸片段,用psti酶切后长度分别为1242bp和3171bp的核酸片段,电泳图如图1所示。构建完成的pbbr1mcs-pr质粒图谱如图2所示。
2、一种重组质粒puc-ci857的构建方法,包括以下步骤:
(1)引物设计:人工设计合成特异性引物序列
cifor:5’-ggtttcttttttgtgctcatagctgtttcctgtgtgaaattg-3’;
cirev:5’-gatcggcaaggtgttctggtagacaagctgtgaccgtctc-3’
pucfor:5’-gagacggtcacagcttgtctaccagaacaccttgccgatc-3’;
pucrev:5’-caatttcacacaggaaacagctatgagcacaaaaaagaaacc-3’
(2)pcr扩增:pcr反应体系配制如表1所示。
pcr程序是98℃预变性30s;98℃变性10s,55℃退火30s,72℃延伸30s,30个循环;最后72℃延伸10min。采用pcp20质粒dna为模板和cifor/cirev引物用以扩增ci857目的片段,而采用puc19质粒dna为模板和pucfor/pucrev引物用以扩增rep-lacz目的片段。所述ci857目的片段长度为772bp,puc19目的片段长度为2329bp。
载体构建:将上述扩增的ci857和puc19目的片段通过琼脂糖凝胶电泳进行分离,并切胶回收。将ci857和puc19目的片段用gibsonassembly的方式进行连接,并将连接产物转化至大肠杆菌dh5α中,涂布在含有100μg/ml的氨苄青霉素lb平板上,于37℃培养18h,挑选克隆于卡那霉素液体培养基中,在37℃培养8h,提取质粒验证,获得质粒puc-ci857。质粒puc-ci857用scai酶切后长度为3019bp的核酸片段,用scai和pcii酶切后长度分别为1373bp和1646bp的核酸片段,如图3所示;构建完成的puc-ci857质粒图谱如图4所示。
3、一种重组质粒pri857的构建方法,包括以下步骤:
(1)引物设计:人工设计合成特异性引物序列
pkfor:5’-cagtgaggcacctatctcagtcagaagaactcgtcaagaag-3’;
pkrev:5’-gaaacctgtcgtgccagctgacgttaaatctatcaccgcaag-3’
pcfor:5’-cttgcggtgatagatttaacgtcagctggcacgacaggtttc-3’;
pcrev:5’-cccatagttcatcagttaaccagaacaccttgccgatc-3’
dblcfor:5’-gatcggcaaggtgttctggttaactgatgaactatggg-3’;
dblcrev:5’-cttcttgacgagttcttctgactgagataggtgcctcactg-3’
(2)pcr扩增:pcr反应体系配制如表1所示。
pcr程序是98℃预变性30s;98℃变性10s,55℃退火30s,72℃延伸30s,30个循环;最后72℃延伸10min。采用pbbr1mcs-pr质粒dna为模板和pkfor/pkrev引物用以扩增pr-kanr目的片段,采用puc-ci857质粒dna为模板和pcfor/pcrev引物用以扩增ci857目的片段,采用pkv6质粒dna为模板和dblcfor/dblcrev引物用以扩增dbl-cole1目的片段。所述plac-ci857目的片段长度为937p,pr-kanr目的片段长度为935bp,dbl-cole1目的片段长度为1679bp。
载体构建:将上述扩增的plac-ci857,pr-kanr和dbl-cole1目的片段通过琼脂糖凝胶电泳进行分离,并切胶回收。将plac-ci857,pr-kanr和dbl-cole1目的片段用gibsonassembly的方式进行连接,并将连接产物转化至大肠杆菌dh5α中,涂布在含有100μg/ml的氨苄青霉素lb平板上,于37℃培养18h,挑选克隆于卡那霉素液体培养基中,在37℃培养8h,提取质粒验证,获得质粒pri857。
琼脂糖凝胶电泳参见《分子克隆》中琼脂糖凝胶电泳的方法,胶回收采用qiagen胶回收试剂盒,化学转化法参见《分子克隆》中氯化钙制备和感受态转化大肠杆菌的方法。连接体系和方法参见gibsonassembly。质粒经kpni酶切后的大小为3430bp的核酸片段,经psti酶切后的大小分别为1952bp和1478bp的核酸片段,验证图如图5所示。构建完成的pri857质粒图谱如图6所示。将dh5α/pri857涂布在卡那霉素50μg/ml的固体培养基,分别于37℃和30℃培养,结果如图7所示。
实施例二:一种零背景克隆载体pri857用于克隆gfp基因的应用
一种零背景克隆载体pri857用于克隆gfp基因的应用,包括以下步骤:
(1)细菌培养:在卡那霉素50μg/ml的液体培养基中接种含有质粒pri857的dh5α,于37℃培养8h;在卡那霉素50μg/ml的液体培养基中接种含有质粒ptd103luxi-sfgfp的dh5α,于37℃培养8h。分别提取pri857和ptd103luxi-sfgfp两种质粒。
(2)引物设计:人工设计合成特异性引物序列
gfpfor:5’-gtcacactattgtatcgctg-3’
gfprev:5’-taagcttttacgctgcaagg-3’
(3)pcr扩增:pcr反应体系是5×q5pcrbuffer10μl,5×enhancer10μl,dntp(10mmol/l)1μl,引物(10μmol/μl)各2.5μl,质粒dna(约20ng/μl)1μl,q5dna聚合酶(5u/μl)0.5μl,加h2o至50μl。pcr程序是98℃预变性30s;98℃变性10s,55℃退火30s,72℃延伸30s,30个循环;最后72℃延伸10min。采用ptd103luxi-sfgfp质粒dna为模板和gfpfor/gfprev引物,用以绿色荧光蛋白基因,该目标dna片段为880bp。
(4)载体酶切:将pri857质粒dna用限制性内切酶bfrbi进行酶切,胶回收线性化片段pri857。
(5)平末端连接:将线性化pri857与gfp片段以摩尔比1:3的比例进行连接,在t4连接酶的作用下,于16℃连接1h,然后转化到dh5α化学感受态细胞中,于30℃培养12h即可。
试验结果表明:通过ci857阻遏基因来控制pr启动子下游kanr基因的表达,只有成功插入gfp目的片段的克隆可以在30℃的培养条件下存活,而自连的载体会因为不能表达卡那霉素抗性蛋白而凋亡,克隆结果可见克隆平板,每个克隆都是绿色的,结果如图8所示。
实施例三:一种零背景克隆载体pri857用于基因组文库构建的应用
一种零背景克隆载体pri857用于基因组文库构建的应用,包括以下步骤:
(1)细菌培养:在卡那霉素50μg/ml的液体培养基中接种含有质粒pri857的dh5α,于37℃培养10h;同时在lb液体培养基中接种蜡状芽孢杆菌,于37℃培养10h。分别提取pri857质粒和蜡状芽孢杆菌基因组dna。
(2)载体酶切:将pri857质粒dna用限制性内切酶bfrbi进行酶切,通过琼脂糖凝胶回收线性化pri857dna片段。
(3)基因组dna酶切:将基因组dna用限制性内切酶alui进行部分酶切,通过琼脂糖凝胶回收dna片段。
(4)平末端连接:将线性化pri857与经alui部分酶切的蜡状芽孢杆菌基因组dna以摩尔比1:3的比例进行连接,在t4dna连接酶的作用下,于16℃过夜连接,然后转化到dh10b电转化感受态细胞中,于30℃培养18h。
试验结果表明:通过ci857阻遏基因来控制pr启动子下游kanr基因的表达,只有成功插入目的片段的克隆可以在30℃的培养条件下存活,而自连的载体会因为不能表达卡那霉素抗性蛋白而凋亡,挑取10个克隆提质粒,通过bamhi酶切如图9所示。
- 还没有人留言评论。精彩留言会获得点赞!