一种马来酸酐熔融接枝聚烯烃材料的制备方法与流程
本发明属于高分子材料领域,具体涉及高接枝率、高分子量或高接枝率、低交联度的马来酸酐熔融接枝聚烯烃材料的制备方法。
背景技术:
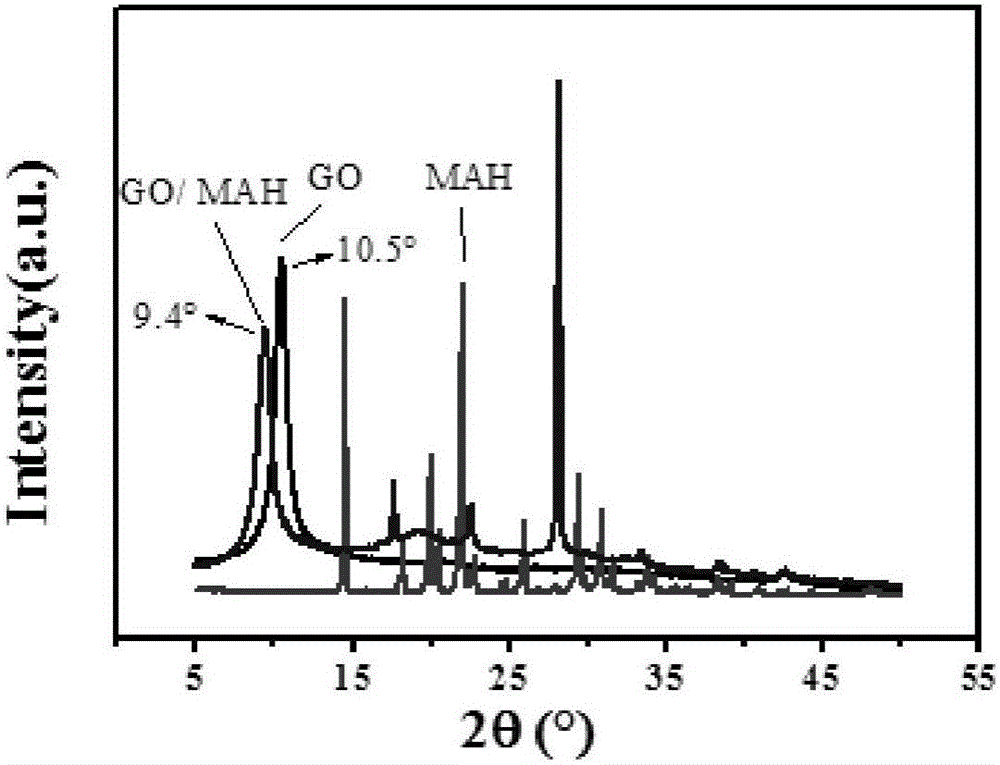
:聚烯烃(PO,polyolefin)是一种具有优异综合性能的通用塑料,广泛应用于汽车、电器、包装等领域。但由于聚烯烃是非极性聚合物,与极性聚合物(尼龙、聚乳酸等)、极性填料(玻纤、碳纤等)不相容。为了提高聚烯烃的极性,通常加入过氧类引发剂和乙烯类不饱和极性单体。工业生产中,为了获得较高的接枝率,通常要在聚烯烃中添加高含量的过氧化二异丙苯(DCP)和马来酸酐(MAH),如添加0.5phr(重量百份数,聚烯烃含量为100份)的过氧化二异丙苯和5phr的马来酸酐,虽然此时接枝率提高了,但由于过氧化二异丙苯含量较高,聚烯烃出现了严重的副反应:丙烯型聚烯烃出现降解(如聚丙烯PP),乙烯型聚烯烃出现交联(如聚乙烯PE)。为了抑制副反应,并同时提高接枝率,通常加入苯乙烯(St)这一富电子类型的第二单体,即PO/DCP/MAH/St体系。然而该体系也存在一定的局限性,比如聚丙烯(PP,polypropylene)体系中190℃下固定马来酸酐含量为5phr,引发剂含量较低时(≤0.3phr),苯乙烯抑制降解的作用有限,引发剂含量较高时(≥0.5phr),苯乙烯的加入反倒增加了降解,又比如聚乙烯(PE,polyethylene)体系PE/DCP/MAH/St体系主要的副反应是聚乙烯的交联,而苯乙烯对聚乙烯交联的抑制作用同样有限。因此难以得到同时具有高接枝率、高分子量或高接枝率、低交联度的马来酸酐接枝聚烯烃PO-g-(MAH-co-St)材料。为了抑制PO/DCP/MAH/St体系的降解或交联,本发明加入氧化石墨烯(GO)这一新型填料。熔融共混过程中,由于π-π作用(马来酸酐上的碳碳双键和氧化石墨烯上类苯环sp2杂化结构组成的大的π电子共轭体系)和氢键作用(马来酸酐上的强极性羰基基团与氧化石墨烯上的羧基、羟基等极性基团),马来酸酐能够插入氧化石墨烯层间,使其层间距变大,从而形成氧化石墨烯-马来酸酐复合物。该复合物能够淬灭聚丙烯叔碳链段自由基(PP3·)、聚乙烯仲碳链段自由基(PE2·),即抑制前者发生β-断裂,也就抑制了聚丙烯的降解,抑制后者发生交联,也就抑制了聚乙烯的交联。但由于苯乙烯共轭体系中丰富的电子云,PP3~St·和PE2~St·活性较高,该复合物并不能有效地淬灭这两种连接苯乙烯的链段自由基,因而这两种自由基能够有效地进行接枝反应。这样,较高引发剂含量下(0.3phr~3phr)聚烯烃及其衍生物体系中的降解或交联仍能够得到有效抑制,但PP3~St·或PE2~St·与马来酸酐单体的接枝过程并不能被有效抑制,因而可以得到同时具有高接枝率、高分子量或高接枝率、低交联度的马来酸酐-苯乙烯共接枝聚烯烃材料PO-g-(MAH-co-St)。技术实现要素:本发明的目的是提供一种马来酸酐熔融接枝聚烯烃材料的制备方法,其接枝率高、分子量高或接枝率高、交联度低。为了实现上述目的,本发明马来酸酐熔融接枝聚烯烃材料的制备方法,具体步骤如下:步骤(1)、将重量份数为0.01~15份的马来酸酐、重量份数为0.01~10份的氧化石墨烯直接加入到重量份数为100份的聚烯烃粒料或粉料上,常温下混合均匀,将该物理混合物加入到反应设备;步骤(2)、将重量份数为0.01~5份的引发剂、重量份数为0.01~10份的苯乙烯液体混合均匀,将该混合物加入反应设备,在一定温度下熔融共混而成;其中所述的反应设备为密炼机、反应型挤出机等,反应温度区间为160~230℃,其中密炼机转子转速为50~150rpm,反应型挤出机螺杆转速为30~600rpm。所述的聚烯烃为聚丙烯、聚乙烯、以及聚丙烯衍生物和聚乙烯衍生物,其中聚丙烯衍生物为乙烯-丙烯嵌段共聚物、乙烯-丙烯无规共聚物,聚乙烯为低密度聚乙烯,高密度聚乙烯等。所述的引发剂为过氧化物类引发剂,即过氧化二异丙苯、双叔丁基过氧异丙基苯、叔丁基过氧化氢、过氧化叔丁基苯甲酸酯、过氧化叔丁基二碳酸酯、2,5-二甲基-2,5-双(叔丁基过氧基)己烷、、2,5-二甲基-2,5-双(叔丁基过氧基)己炔、过氧化苯甲酰中的一种或几种。所述的氧化石墨烯为单层或多层氧化石墨烯粉末,即纯度为80%~99%,厚度为0.34~17nm,层数为1~50层,片层直径为5~100μm,比表面积为50~1000m2/g,褐黄色或黑色粉末,表面及边缘主要为羧基、羟基、环氧基团,可吨级量产的单层或多层氧化石墨烯。与现有技术相比,本发明的优点和有益效果如下:(1)本发明制备的马来酸酐接枝聚烯烃接枝率高、分子量高,或是接枝率高、交联度低;(2)本发明与马来酸酐-苯乙烯共单体接枝聚烯烃体系相比,由于氧化石墨烯的加入可有效抑制聚丙烯叔碳链段自由基的降解、聚乙烯仲碳链段自由基的交联,较高的引发剂含量并不会引起较大降解或交联,可实现降解和交联的有效控制或调节;(3)本发明首次将氧化石墨烯应用于马来酸酐熔融接枝聚烯烃体系中,不同于以往氧化石墨烯在复合材料、药物负载等体系的应用,本发明拓展了氧化石墨烯的应用范围,为氧化石墨烯的大规模应用提供了新的可能。(4)本发明制备的马来酸酐接枝聚烯烃不同于普通的增容剂,该增容剂中存在氧化石墨烯,具有较高的热稳定性、气体阻隔性能等;(5)本发明在马来酸酐接枝聚烯烃增容剂制备过程中完全不涉及溶剂的使用,所有组分并不需要进一步处理,直接通过搅拌机进行物理混合;(6)本发明通过密炼机或挤出机熔融共混制备所需增容剂,该方法具有工业大规模生产的可能。附图说明图1为GO、GO-MAH、MAH的红外图谱;图2为GO、GO-MAH、MAH的X射线衍射图谱;图3为GO、GO-MAH、MAH的热失重稳定曲线的重量-温度图谱;图4为GO、GO-MAH、MAH的热失重稳定曲线的失重速率-温度图谱;图5为制备得到的实施例1和比较例1样品照片。具体实施方式下一步结合实施例,进一步说明本发明:实施例中为了测定马来酸酐接枝聚烯烃的接枝率,首先对PO-g-MAH接枝物进行纯化,然后采用红外光谱和酸碱滴定结合的测定方法来表征接枝率。PO-g-MAH纯化的具体步骤如下:首先,取1.5g样品加入50ml二甲苯中,140℃油浴30min,样品完全溶解,趁热将溶液倒入100ml丙酮中,然后将该滤液抽滤,抽滤物70℃真空干燥3h;其次,将干燥过的抽滤物用滤纸包裹起来放入丙酮抽提装置,90℃油浴下抽提24h;最后,将抽提物70℃真空干燥8h,得到干燥的纯化样品。傅里叶红外光谱法(FT-IR)的具体步骤如下:首先,取0.3g左右纯化样品置于聚四氟乙烯膜上,平板硫化仪中190℃下预热5min,60MPa下热压2min,通水冷却至常温取出,轻轻将所压薄膜揭起;其次,选择100μm厚度左右的薄膜进行红外测试:透过模式TR,吸光度absorbance,分辨率1cm-1,扫描范围:4000-400cm-1,扫描次数64scans;最后,使用omnic软件分别对1780cm-1、2721cm-1、1470cm-1峰进行积分,得到峰面积A1780、A2721和A1470,A1780/A2721、A1780/A1470即为吸光度之比Ra,可定量来表征接枝率的大小。其中1780cm-1为酸酐中的羰基峰,2721cm-1为聚丙烯链段骨架峰,1470cm-1为聚乙烯亚甲基特征峰,即内标峰。酸碱滴定纯化样品的具体步骤如下:首先,取0.5g左右纯化样品放入装有50ml二甲苯的三颈烧瓶(容积250ml)中,油浴温度150℃,加热回流20min,样品充分溶解;其次,将油浴温度调至100℃,并使三颈烧瓶悬空于油浴装置,待三颈烧瓶中二甲苯溶液冷却至70℃,磁子搅拌溶液的同时缓慢滴入10ml氢氧化钾-异丙醇溶液,此时将三颈烧瓶置于油浴中100℃加热回流1.5h,使羧基化的马来酸酐与氢氧化钾充分反应;然后,冷却油浴至80℃,此时溶液温度70℃,趁热滴加5滴酚酞溶液,溶液呈粉红色;最后用盐酸-异丙醇溶液趁热滴定至无色,该过程中由于滴入的盐酸-异丙醇溶液是常温的,所以滴定过程中的二甲苯溶液温度会降低,可在滴定过程中适当升高油浴温度来保持二甲苯溶液温度为70℃。重复该实验3次,计算出接枝率G1。空白样(不加纯化样品)的滴定过程同上,计算出接枝率G2。最终,酸碱滴定法得到的纯化样品的接枝率为,G=G1-G2。熔融指数测定按照GB3682-83标准在高铁熔融指数仪上进行测试:230℃,2.16kg。下面结合实施例对本发明进行详细说明。然而,这些实施例仅作为示例性目的,本发明的范围不限于此。实施例1:PP/DCP/MAH/St/GOPP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例1:PP/DCP/MAH/StPP/DCP/MAH按照重量比100/0.5/5(其中聚丙烯称量46g)进行混合并进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例2:PP/DCP/MAH/GOPP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例3:PP/DCP/St/GOPP/DCP/GO按照重量比100/0.5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例4:PP/DCP/MAHPP/DCP/MAH按照重量比100/0.5/5(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例5:PP/DCP/StPP/DCP按照重量比100/0.5(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例6:PP/DCP/GOPP/DCP/GO按照重量比100/0.5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例7:PP/DCPPP/DCP按照重量比100/0.5(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例8:纯PP按照46g聚丙烯固体颗粒加入密炼机中,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。实施例2:PP/DCP/MAH/St/GO(改变DCP含量为1.0phr)PP/DCP/MAH按照重量比GO/100/1.0/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例9:PP/DCP/MAH/GO/100/0.5/5/0.1PP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。实施例3:PP/DCP/MAH/St/GO(改变温度为180℃)PP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,180℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例10:PP/DCP/MAH/100/0.5/5,180℃PP/DCP/MAH按照重量比100/0.5/5(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,180℃下以50rpm的速度混炼5min。样品的测试结果见表1。比较例11:PP/DCP/MAH/GO/100/0.5/5/0.1PP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。实施例4:PP/DCP/MAH/St/GO,180℃PP/DCP/MAH/GO按照重量比100/3.0/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,180℃下以50rpm的速度混炼5min。样品的测试结果见表1。实施例5:PP/DCP/MAH/St/GOPP/DCP/MAH/GO按照重量比100/3.0/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表1。实施例6:PP-g-MAH的热失重稳定性。可以看出,PP/DCP/MAH/100/0.5/5(190℃)中加入0.1phr的GO之后,T5%提高了11℃,Tmax降低了2.5℃。样品的测试结果见表2。实施例7:PE/DCP/MAH/St/GOPE/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表3。比较例12:PE/DCP/MAH/StPE/DCP/MAH按照重量比100/0.5/5(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,而后再加入2.5phr的液体状St,190℃下以50rpm的速度混炼5min。样品的测试结果见表3。比较例13:PE/DCP/MAHPE/DCP/MAH按照重量比100/0.5/5(其中聚丙烯称量46g)进行混合并手动搅拌均匀,加入密炼机中,190℃下以50rpm的速度混炼5min。样品的测试结果见表3。实施例8:GO、GO-MAH、MAH的红外图谱。详见图1。其中GO-MAH红外样品制备方法如下:分别称量39mg的GO粉末、28mg的块状MAH,使用研钵研磨,研磨过程中滴加3滴丙酮;40℃真空干燥12h后,该复合物出现块状物,重新研磨。GO、MAH两个样品与GO-MAH样品经历了相同的制备过程,研磨过程中滴加丙酮,然后干燥。选取少量的干燥样品,使用溴化钾压片法压片,透射模式,对制备样品进行红外测试(经过对比,未经处理与研磨-干燥处理的的GO、MAH样品的红外图谱相同,排除了研磨-干燥处理过程对GO、MAH样品的影响)首先,相比于GO,GO/MAH的变化如下,(1)GO/MAH中的羟基“OH“峰向低波数偏移,且峰形钝化,这可能是由于氢键的存在(酸酐与GO上的羧基、羟基之间);(2)GO中存在大的吸收峰:3683-2814cm-1,GO/MAH中的对应吸收峰范围变大很多:3683-1860cm-1,这可能是由于GO中的不饱和六元碳环π-π共轭结构与MAH中碳碳双键π-π共轭结构发生共轭,从而使1780cm-1、1860cm-1等羰基吸收峰消失,3000-2000cm-1范围的碳氢峰波长范围变大;(3)羰基峰略向高波数偏移,1706cm-1到1717cm-1,且强度相对碳碳双键变弱,可能是由于酸酐中的高波数羰基吸收峰的影响,即与GO中的羰基发生氢键作用,因而向高波数偏移,同时由于GO与MAH间的共轭作用,羰基峰强度相对碳碳双键变弱;(4)“C-OH”峰消失,可能是由于MAH与其反应;其次,相比于MAH,GO/MAH的变化如下,GO-MAH中1780-1860cm-1的羰基伸缩振动峰消失,只剩下1717cm-1的羰基吸收峰,这可能是由于GO与MAH之间的共轭效应、氢键效应,使其电子云密度平均化。样品的测试结果见图1。实施例9:X射线衍射,GO、GO/MAH、MAH的XRD样品使用实施例8中红外制备样品。详见图2。根据布拉格方程2dsinθ=nλ计算可知,氧化石墨烯与马来酸酐研磨以后,层间距由0.77nm变为0.94nm,这表明马来酸酐插入氧化石墨烯层间,表明二者间由明显的相互作用,与红外结果对应,表明熔融接枝过程中确实生成了GO/MAH复合物,正是该复合物抑制了其降解、接枝。样品的测试结果见图2。实施例10:热失重稳定性曲线,GO、GO/MAH、MAH的XRD样品使用实施例8中红外制备样品。GO-MAH的热稳定性下降,GO-MAH在100℃附近出现新的Tmax特征峰,这应当是由于MAH插入了GO层间,并且插入GO层间的MAH的热稳定性低于纯的MAH,这表明马来酸酐插入氧化石墨烯层间,表明二者间由明显的相互作用。样品的测试结果见表1、图3和图4。实施例11:PP-g-MAH(实施例1和比较例1)样品的图片和SEM图。图5中可以看出,片状的比较例1样品为乳白色,而片状的实施例1样品为灰色,虽然有少量氧化石墨烯发生团聚形成黑色小颗粒,但总的来说氧化石墨烯还是均匀分散于其中的。样品的测试结果见表1和图5。表1.马来酸酐接枝聚丙烯体系的熔指和吸光度之比SampleT5%(℃)Tmax(℃)NeatPP355426.4PP/DCP/MAH/100/0.5/5(190℃)398468.2PP/DCP/MAH/GO/100/0.5/5/0.1(190℃)409465.7PP/DCP/MAH/GO/100/0.5/5/0.1(190℃)412466.1表2.PP-g-MAH的热失重稳定性表3.马来酸酐接枝聚乙烯体系的熔指和吸光度之比实施例12:将5gMAH、0.05gGO直接加入到50g乙烯-丙烯嵌段共聚物粒料中,将该混合物搅拌均匀后加入到密炼机;将0.005g双叔丁基过氧异丙基苯、0.005gSt混匀后加入到上述密炼机中,160℃下以100rpm的速度混炼5min。实施例13:将0.005gMAH、0.5gGO直接加入到50g乙烯-丙烯嵌段共聚物粒料中,将该混合物搅拌均匀后加入到密炼机;将2.5g叔丁基过氧化氢、0.5gSt混匀后加入到上述密炼机中,230℃下以60rpm的速度混炼5min。实施例14:将7.5gMAH、1gGO直接加入到50g等规聚丙烯粒料中,将该混合物搅拌均匀后加入到螺杆挤出机;将1g过氧化叔丁基苯甲酸酯、2.5gSt混匀后加入到上述螺杆挤出机中,180℃下以200rpm的速度挤出。实施例15:将0.05gMAH、5gGO直接加入到50g乙烯-丙烯无规共聚物粒料中,将该混合物搅拌均匀后加入到螺杆挤出机;将0.05g过氧化叔丁基苯二碳酸酯、5gSt混匀后加入到上述螺杆挤出机中,170℃下以400rpm的速度挤出。实施例16:将500gMAH、5gGO直接加入到5000g高密度聚乙烯粒料中,将该混合物搅拌均匀后加入到螺杆挤出机;将5g过氧化叔丁基苯二碳酸酯、500gSt混匀后加入到上述螺杆挤出机中,170℃下以400rpm的速度挤出。实施例17:将50gMAH、1gGO直接加入到5000g高密度聚乙烯粒料中,将该混合物搅拌均匀后加入到螺杆挤出机;将0.2g过氧化叔丁基苯二碳酸酯、0.3g过氧化叔丁基苯甲酸酯、50gSt混匀后加入到上述螺杆挤出机中,230℃下以100rpm的速度挤出。上述实施例并非是对于本发明的限制,本发明并非仅限于上述实施例,只要符合本发明要求,均属于本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 马来酸酐直接接枝聚丙乙交酯制备高分子量mplga的技术的制作方法
- 一种led封装用马来酸酐接枝聚苯醚改性环氧树脂复合材料及其制备方法
- 一种马来酸酐接枝苯乙烯嵌段共聚物共混改性聚氨酯热熔胶及其制备方法
- 一种led封装用高透光率的马来酸酐接枝聚苯醚改性环氧树脂复合材料及其制备方法
- 一种led封装用环保耐候的马来酸酐接枝聚苯醚改性环氧树脂复合材料及其制备方法
- 一种提高led亮度的封装用马来酸酐接枝聚苯醚改性环氧树脂复合材料及其制备方法
- 无机硅-马来酸酐接枝聚乙烯醇材料及其制备方法与应用
- 马来酸酐接枝改性大豆蛋白生物胶黏剂及其制备方法
- 一种聚丙烯/马来酸酐接枝聚丙烯/细菌纤维素复合材料及其制备方法
- Rgd多肽接枝聚(马来酸酐-己二胺-dl-乳酸)/改性羟基磷灰石多孔复合材料的制备方法