一种分段扩增猪流行性腹泻病毒全基因组的引物组及方法与流程
本发明属于生物技术领域,具体地,涉及一种分段扩增猪流行性腹泻病毒全的基因组的引物组及方法。
背景技术:
猪流行性腹泻的病原为猪流行性腹泻病毒(porcine epidemic diarrhea virus,PEDV),其主要诱发猪只肠道疾病,其症状表现为呕吐、腹泻,食欲下降,脱水,且具有显著的日龄依赖现象。特别对于7日龄内的新生仔猪,在缺乏有效母源抗体下,死亡率可高达100%。2010年底,我国养猪场大范围爆发了PED,且近年来仍未得到有效控制对我国养猪业造成了巨大的损失。2013年4月美洲国家陆续爆发PED疫情,随后亚洲其他国家和部分欧洲国家也相继爆发PEDV疫情,再次给世界范围内的养猪业带来了巨大的危害。基于近年流行野毒株的出现及大流行,传统商品疫苗已经对变异毒株缺乏产生充分的抵抗作用。由此,对新流行的PEDV变异株的研究及分析其分子流行病学,可为揭示新发PEDV感染致病的分子机制和特异性诊断的研制提供必要的理论参考依据。
猪流行性腹泻病毒在组成结构上为具有囊膜,且囊膜上有花瓣样纤突,核酸类型为线性,单股正链,不分节段的RNA病毒。在所有RNA病毒中,冠状病毒的基因组长度都相对较长。而PEDV完整的基因组核苷酸约有28,038nt,其基因组序列类似于mRNA一样,5’端具有帽子结构,3’端拥有Poly(A)尾巴。从5’端到3’端,基因结构顺序依次为5’UTR-Replicase(ORF1a/b)-S-ORF3-E-M-N-3’UTR;主要编码的结构蛋白有纤突蛋白(Spike,S)、膜蛋白(Membrane,M)、包膜蛋白(Envelope,E)和核衣壳蛋白(Nucleocapsid,N)。而非结构蛋白有复制酶多聚蛋白(Open Reading Frame 1,ORF1a/b)和ORF3蛋白(Open Reading Frame 3,ORF3)。PEDV基因组5′端非翻译区(5′UTR)长296nt,包含一个长约65nt-98nt前导序列(L),Kozak序列(GUUCAUGC)和一个短小的开放阅读框(ORF)。3′UTR长度约为334nt,拥有一个所有冠状病毒都有,但位置不同的保守8碱基(GGAAGAGC)序列。PEDV的复制酶多聚蛋白ORF1a/b(pp1ab)由两个亚单位蛋白ORF1a和ORF1b共同组成。在核苷酸序列中,ORF1a/b全长约20345nt,由包含46nt重叠序列的且长度分别为12354nt和8037nt的两个大阅读框组成,占据基因组约2/3的核苷酸数量。ORF1a/b非结构蛋白在病毒感染早期可执行多种重要的生物学功能,其中ORF1a主要执行蛋白切割作用及参与RNA的复制等,而ORF1b主要编码RNA依赖性RNA聚合酶。冠状病毒的纤突蛋白S共同拥有的包括附着于靶细胞,介导囊膜与细胞膜融合等的基本生物学功能。经典毒株CV777(AF353511)的S蛋白由约4152nt编码的1383个氨基酸组成。S蛋白属于1型的膜糖蛋白,从N到C端,分别是信号肽区(1-24aa)、长的细胞外区(25-1333aa)、(1334-1356aa)和较短的细胞质尾部(1357-1383aa)。同时根据其它冠状病毒S蛋白的同源性,还可将S蛋白分为通过折叠锚定的纤突亚蛋白S1(l-789aa)和S2(790-1383aa)。其中S1蛋白还可以再细分为具有结合受体的功能区域S1-NTD(N-terminal domain,19-252aa)和S1-CTD(C-terminal domain,509-638aa)。到目前为止,研究人员在S蛋白上鉴定出1个诱导中和抗体产的区域COE(99–638aa)和3个诱导中和抗体产生的表位,分别是SS2(748YSNIGVCK 755),SS6(764LQDGQVKI 771);2C10,1368GPRLQPY 1374。研究表明,PEDV在细胞内的繁殖需要蛋白水解酶的协助,并且该过程可能涉及S蛋白。当病毒附着于宿主细胞的细胞膜之后,在外源的胰蛋白酶作用下,S蛋白被酶解断裂为两个亚基(S1和S2),使被侵染的细胞发生更明显的病变。此外,S蛋白与体外培养病毒适应能力以及病毒滴度的衰减有关系。ORF3是PEDV惟一辅助基因,其完整的ORF3基因有675nt编码225aa。而在体外细胞传代过程中,ORF3核苷酸可以出现不同样式的缺失,充分表现了其多样性,与病毒毒力有关。在经典疫苗CV777和韩国DR13疫苗毒株中,ORF3基因出现了的49nt的缺失可以作为PCR诊断中,区分PEDV野毒与细胞致弱毒株的基因标记。2012年,Wang等首次发现ORF3蛋白和SARS-CoV的3a蛋白一样,能够在细胞表面形成钾离子通道,提高病毒的滴度。PEDV的E基因由231nt组成,编码76个氨基酸。该蛋白在病毒包膜和病毒粒子的组成等方面起着关键的作用。然而,在DR13致弱毒株中,存在21nt的缺失,E基因在PEDV的致病性和结构作用上仍有待研究。研究发现,E蛋白可以定位于内质网和核仁表面,引起内质网应激效应,不能影响肠道细胞IEC的细胞复制周期,但是可以上调白介素8和细胞凋亡Bcl-2的表达。M基因全长681nt,在目前研究中,其不存碱基的缺失,序列保守性仅次于N基因。M蛋白作为PEDV主要的囊膜结构蛋白,其在病毒粒子的装配和出芽过程发挥着重要作用。Laude,H.在其研究中发现,TGEV在细胞感染中,可以激发机体产生α-干扰素(IFN-α)。M蛋白还可以刺激机体产生中和抗体。除此之外,张志榜等还鉴定了M基因的线性表位195WAFYVR200,且该表位不能被TGEV阳性血清识别。PEDV N基因由1326nt组成,编码441个氨基酸。在PEDV基因组中,在核苷酸数量和种类上,保守性最高。N蛋白作为一种RNA结合蛋白,能与感染细胞中细胞核磷蛋白共定位于细胞核,根据吕茂杰报道,其发现N基因核仁定位信号和N蛋白核仁定位机制提供了依据。因其富含碱性氨基酸,如赖氨酸和精氨酸残基等而表现出高度的碱性,在pH=9.0的低渗非离子溶剂中有最大的溶解度。在冠状病毒已知结构蛋白中,只有N蛋白是磷酸化的蛋白。丝氨酸残基被认为是潜在的磷酸化位点,PEDV CH/S毒株的N蛋白总共拥有35个丝氨酸残基,约占其总氨基酸残基数的7.9%,且主要集中在两个碱性区域内(57-190aa和199-391aa)。PEDV N蛋白被证实可以抑制干扰素调节因子3活性和Ⅰ型干扰素的表达,由此影响机体的免疫系统。Xu等研究发现N蛋白可以延长感染细胞的S周期,同时引起内质网应激和上调白介素8的表达。经广泛的流行调查,目前国内外流行的变异PEDV毒株S基因编码区全长4161nt,而CV777作为经典毒株的S基因长度仅为4152nt。其原因主要为变异株在S基因的S1片段有15nt插入和6nt缺失。据Chen等调查,由于进化差异,PEDV的S基因呈现了序列长度和核苷酸差异的多样性。在NCBI数据库的序列中,S基因长度从3359-4255nt,多达11种长度类型。而4161nt的长度的序列同样占绝大部分(约68.4%),且均为2010年后被检测到的,表明目前该种类的毒株具有较明显的进化优势。除此之外,MF3809/2008/South Korea(KF779469),TC-PC177-P2,Tottori2/JPN/2014以及TTR-2/JPN/2014(LC063828)等近年分离得到的变异毒株在S基因中存在大片段的核苷酸缺失,也属罕见。在序列同源性中,变异毒株与CV777毒株均不高,且存在核苷酸的插入和缺失,提示着PEDV变异毒株相对于30多年前的经典毒株表现较快的变异速度。在主要抗原中和位点中,变异株与经典毒株有着多处氨基酸的突变。这些位点的变化可能是目前疫苗株不能对变异毒株产生良好保护的主要原因。然而关于这些突变的位点以及氨基酸的插入和缺失对S结构基因影响是否对其抗原性、毒力等生物功能仍有待进一步探索。除了S基因的变化,在基因组其他位置,众多可缺失的基因位置和数量都有所差异,也丰富了PEDV毒株的生物多样性,从而也使得该病毒的研究存在更多的不确定性。例如在ORF1a/b基因中,在1036-1043位氨基酸中,可有8个氨基酸缺失(Attenuated DR13等毒株);再者在E基因中,67–87位核苷酸位置上,存在21核苷酸缺失(Attenuated DR13毒株);在ORF3基因中,在246位核苷酸开始,存在49个核苷酸的缺失等。
PEDV作为单股RNA类病毒,相比DNA类病毒存在更高的突变概率。虽然从90年代就开始应用疫苗免疫PED,但2006年Chen等报道有少量即使免疫了二联疫苗的猪场仍然会发生PED疫情。直到2010年底,PED疫情在我国大面积流行,这表明PEDV毒株一直在不断变异,现行疫苗对其的抵抗作用越来越小。据目前报道,体外试验中变异毒株与CV777毒株的血清学反应结果是不高的。虽然近年的G2亚群毒株仍然不断扩散,但是自从2006年以后,G1亚群毒株被报道的频率远远少于G2亚群,这应是CV777为基础的疫苗发挥了很大的作用。所以,获得足够的PEDV基因信息,对防控和诊治PED的疫情将提供关键的诊断基础。
技术实现要素:
本发明所要解决的技术问题是克服现有技术中猪流行性腹泻病毒基因信息少,难以防控和诊治的缺陷和不足,提供一种分段扩增猪流行性腹泻病毒全基因组的引物组。本发明通过设计22对引物组对PEDV进行全覆盖式地扩增,更便利地扩增出PEDV全长基因,将庞大的基因组细分为多段,可便于定点研究特定区段的核苷酸变化,包括位点突变、核苷酸插入与缺失等,以更好把握PEDV基因的变化动态,了解毒株的变化,为解决该病提供准确的分子信息。
在该方案中,本发明首要需克服的技术问题是如何设计匹配引物组,能够在实现扩增PEDV全长基因信息目的得基础上,能够将PEDV的引物扩增片段长度控制在约1600bp以内,以适应当前相对比较成熟且廉价的第一代基因测序技术,且由引物扩增得到的序列片段间可以首尾相连,且有足够的重叠序列(35~931bp)。以及摸索设计出来的22对引物各自的最佳退火温度和时间,探索出最佳的PCR体系和程序。
本发明的目的是提供一种分段扩增猪流行性腹泻病毒全基因组的引物组。
本发明的另一目的是提供利用上述引物组扩增猪流行性腹泻病毒全基因组的方法。
本发明的上述目的是通过以下技术方案给予实现的:
一种分段扩增猪流行性腹泻病毒全基因组的引物组,所述引物组包括22个引物对,各引物对上下游引物依次如如SEQ NO.1~SEQ NO.44所示。
本发明根据GenBank数据库中CV777(AF353511)的核苷酸序列,利用生物信息学方法,设计并筛选了用于扩增PEDV全长基因组核苷酸信息的22对引物,运用分子生物学手段成功扩增出来了22对扩增产物,利用基因测序技术和生物信息学技术,获得PEDV全长基因信息。而所述PEDV基因组的5’和3’末端都采用了小片段扩增设计,使扩增产物控制在700bp内,主要是兼顾了要使用非常规的引物扩增5’UTR前端和3’UTR末端序列,如采用SMARTerTMRACE cDNA Amplification扩增技术扩增5’UTR最前端和3’UTR最末端准确的核苷酸序列。本发明设计的引物组相邻两个扩增片段之间具有39~931bp的重叠同源序列,便于测序拼接,同时所有引物组的扩增片段均小于1600bp,易于PCR反应的进行。
一种分段扩增猪流行性腹泻病毒全基因组的方法,所述方法为:提取PEDV总RNA,反转成cDNA,分别利用上述上述22个引物对进行PCR扩增反应,分别进行克隆测序(将PCR产物连接克隆载体,选取阳性克隆,DNA测序鉴定),最后拼接序列,得到PEDV全基因组序列。
其中,所述PCR产物均小于1600bp,易于PCR反应的进行。
所述PCR扩增反应同时在一个温度梯度PCR仪中进行,通过采用同一个PCR反应程序,不同引物对应不同退火温度,一次性、高效地完成PEDV的全基因组序列扩增。
优选地,所述PCR仪的温度梯度为48℃~57℃。
优选地,所述PCR扩增反应程序为:94℃预变性1min;94℃变性6s,退火温度下(48℃~56.1℃)退火20s,72℃延伸20s,循环36次;72℃终延伸7min;所述退火温度对应为22个引物对各自对应的退火温度。
优选地,所述PCR扩增反应的体系为:上游引物(10μM)2.0μL、下游引物(10μM)2.0μL、cDNA 2.0μL、2×PrimerSTAR Max 25.0μL、ddH2O 19.0μL。
优选地,22个引物对分别为引物对5'、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、3',各引物对上下游引物依次如SEQ NO.1~SEQ NO.44所示;
22个引物对的退火温度分别为:
引物对3'、引物对9、引物对10、引物对11、引物对13、引物对16、引物对20的退火温度均为56.1℃;
引物对1、引物对6、引物对7、引物对8、引物对14、引物对17、引物对18、引物对19的退火温度均为55.2℃;
引物对5'、引物对2、引物对5的退火温度均为54.1℃;
引物对3的退火温度为53.0℃,引物对4的退火温度为50.8℃,引物对12的退火温度为48.9℃,引物对15的退火温度为48.0℃。
另外,具体优选地,上述方法中,所述克隆测序的具体方法为将PCR扩增产物回收纯化,扩增产物3’末端加碱基A,然后连接至pMD18-T载体,转化Top 10感受态细胞,筛选和鉴定重组菌后,进行测序;
所述拼接序列的具体方法为:测序结果利用Lasergene 7.10生物信息学软件中的Editseq软件进行去除包括引物对在内的外向端序列,全长序列的拼接及同源性分析;同时,运用NCBI中GenBank快速比对工具BLAST进行测序结果的验证或比对搜索,完成序列与数据库中的已有序列进行比对分析。
另外,上述引物组或/和扩增方法在猪流行性腹泻病毒基因或基因组扩增方面的应用亦在本发明保护范围内。
同时,本发明还提供一种扩增猪流行性腹泻病毒基因组上特定区域的方法,所述方法为根据需要任意组合上述22个引物对进行PCR扩增反应。
另外,上述方法在猪流行性腹泻病毒基因组特定区域研究上的应用亦在本发明保护范围内。
优选地,所述应用为检测猪流行性腹泻病毒突变位点、核苷酸插入或缺失上的应用。
本发明上述方法是一种操作简易、快速、稳定、分区段地扩增猪流行性腹泻病毒全基因组的方法。首先以PEDV的cDNA为模板,以自行设计的22对引物,通过高保真DNA聚合酶,扩增获得PEDV的全长序列。将22对引物扩增的PCR产物连接克隆载体、选取阳性克隆、DNA测序鉴定,再将所测序列通过软件拼接,得到PEDV全长核苷酸序列信息。本发明主要优点在于可以在一个温度梯度PCR仪器里面,通过采用同一PCR反应程序,不同引物与不同退火温度,一次性、高效地完成PEDV的全基因组序列扩增。由于是全覆盖式地扩增,且扩增片段均不超过1600bp,所以也便于根据所需研究的特定区域进行定区域扩增。
与现有技术相比,本发明具有以下有益效果:
(1)本发明的22对引物对扩增特异性和敏感性好,可对PEDV进行全覆盖式地扩增。且引物组的5’和3’末端都采用了小片段扩增设计(主要是兼顾了如要使用SMARTerTMRACE cDNA Amplification扩增技术扩增5’UTR最前端和3’UTR最末端的核苷酸序列),扩增片段长度控制在1600bp以内,可以适应当前相对比较成熟且廉价的第一代基因测序技术。22对引物设计位点合适,相邻两个扩增片段之间具有重叠同源序列,便于测序拼接。
(2)本发明所述引物组能够实现完美匹配,扩增方法通过在一个温度梯度PCR仪中同时采用相同的PCR反应体系和程序,一次性高效地完成了PEDV的全基因组序列扩增。
(3)由于PEDV全长基因组较大,本发明的引物组和方法将庞大的基因组细分为多段,可便于定点研究特定区段的核苷酸变化,包括位点突变、核苷酸插入与缺失等,可以更好地把握PEDV基因的变化动态,了解毒株的变化,为解决该病提供准确的分子信息。
附图说明
图1为本发明设计的22对引物在PEDV基因组中的位置分布图。
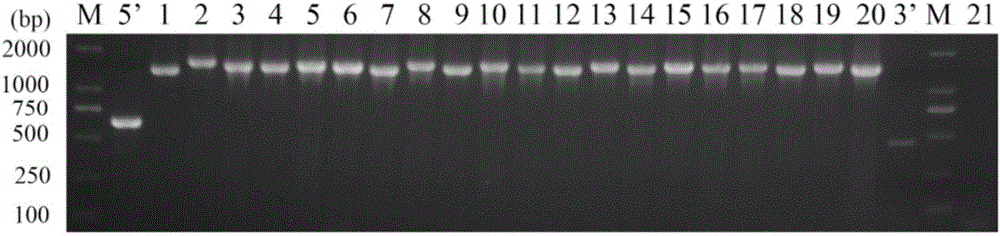
图2为本发明设计的22对引物扩增结果琼脂糖凝胶电泳图。
图3为5’引物对测序片段去除上游引物及其外向端序列过程图。
图4为由参考序列指引的22段去除引物信息的拼接效果图。
具体实施方式
下面结合说明书附图和具体实施例对本发明作出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
实施例1猪流行性腹泻病毒(PEDV)全长引物设计
1、根据GenBank数据库中CV777(AF353511)的核苷酸序列,经过大量的设计筛选研究,最终得出了22对引物用于扩增PEDV全基因组,所述引物组如表1所示,引物对之间重叠的位置及大小如表2所示,引物的最佳退火温度及温度梯度如表3所示。
表1全长引物序列表
注:相对核苷酸位点参考毒株SHQP/YM/2013(GenBank收录号:KJ196348)。
表2全长引物序列重叠长度表
注:引物对14与引物对15重叠区间核苷酸数量为931,主要是优先考虑了引物对16、引物对17、引物对18的组合可以完整地扩增PEDV序列中重点研究序列区间的S基因。而促使引物对15向前推移,从而与引物对14有较多重叠。
表3全长引物最佳退火温度及温度梯度PCR仪设定
注:梯度PCR为8*12孔数,温度梯度设置为(48℃~57℃)。
实施例2猪流行性腹泻病毒(PEDV)全基因组扩增方法
1、PEDV流行毒株的获取
采集临床上疑似感染PEDV的猪只肠道组织样品,低温保存并剪取若干个黄豆大的组织块进行研磨。研磨中混入石英砂,添加约5mL左右预冷的PBS。匀浆收集到10mL Ep管中,置-80℃超低温冰箱中冻融3次。样品再经10,000×g 4℃离心10min。取上清,经0.22μm滤膜过滤。将所得到的研磨滤液分成若干管冻存于-80℃超低温冰箱中,待核酸提取用。
2、组织样品总RNA的提取
用Magen超纯RNA提取试剂盒提取研磨液中总RNA。取滤液250μL加入750μL的Trizol中,取250μL的PBS于灭菌离心管中,加入750μL TRIzol,剧烈振荡,室温下作用5分钟。再加200μL氯仿,剧烈振荡,室温下放置5分钟。4℃12,000r/min离心15分钟。将500μL上层水相移到另一新的灭菌微量离心管中,加入500μL异丙醇,轻柔混匀,-20℃放置15分钟。12,000r/min离心15分钟,倾去上清液。加入1,000μL冰预冷的75%乙醇,缓慢颠倒摇匀。4℃12,000r/min离心15分钟。轻轻弃去上清,放置3-5min以风干沉淀。加入20μL的RNase Free双蒸水溶解RNA,于-20℃保存备用。
3、病毒cDNA第一链的合成
参照东洋纺(上海)生物科技有限公司的改良型高效逆转录酶试剂盒ReverTra Ace(TRT-101)的说明书进行。取提取的RNA进行cDNA第一链的合成,20μL的反转录体系中包括以下各组分(如表4):
表4反转录体系
将以上组分充分混匀,然后立即置于42℃水浴锅中孵育1h,所得到的cDNA置于-20℃保存。
4、PCR扩增方法
运用22对全长引物,使用以下PCR反应体系和反应程序进行扩增(如表5):
表5PCR反应体系
PCR扩增程序:
5、扩增产物的回收与纯化
将所有扩增产物进行琼脂糖凝胶电泳,在紫外灯短暂照射下切取目的片段,参照Magen公司DNA快速回收试剂盒进行回收纯化,具体方法如下:
(1)将目的基因DNA片段经0.8~1%的琼脂糖凝胶电泳分离;
(2)电泳结束后,将琼脂糖凝胶经EB染色后,在长波紫外灯下用洁净、锋利的手术刀片切取含目的基因片段的琼脂凝胶块,放入一个洁净的1.5或2mL离心管中;
(3)按1:3的比例(凝胶质量:溶胶液体积(mg:mL)加入溶胶液(Extraction Buffer);
(4)于56℃恒温水浴或金属洛中温育10-15min,直到凝胶块融化,温育过程中每隔2~3min上下颠到混匀一次;
(5)将混合液转移至回收柱内,6,000r/min,离心1min,弃去管内液体;
(6)将500μL溶胶液加入回收柱内,12000r/inin,离心1min,弃去管内液体;
(7)向回收柱内加入750μL洗涤液(Wash Buffer),室温静置2~5min,12,000r/min,离心1min,弃去管内液体;
(8)再次于12,000r/min,离心2min,然后将回收柱转移到无菌1.5mL离心管中,打开盖子,室温静置2~5min,待其自然风干;
(9)向离心柱内加30~50μL洗脱液、ddH2O或TE溶液,于37℃温育5min;
(10)12,000r/min,离心3min,如有需要可将离心管中的液体再次加入到离心柱内,再次洗脱,离心管底部的液体即为回收的目的基因片段,可直接用于连接反应,或于-20℃保存备用。
6、扩增产物3’末端加碱基A
由于高保真酶扩增出来的PCR片段为平末端,而普通Taq酶具有在PCR产物3’末端加一个碱基A的特性,由此可以与粘性末端的pMD18-T载体连接,从而可以大大提高片段与载体连接的效率。具体反应体系如表6:
表6扩增产物3’末端加碱基A反应体系
反应程序为72℃30min。
7、PCR片段与载体连接
将3’末端加碱基A的PCR产物进行浓度测定,并以1:3/1:5的摩尔比例与pMD18-T载体进行连接(连接体系如表7)。
表7连接体系
上述体系经充分混匀后,置于连接仪中,16℃连接过夜。次日将连接产物放入65℃水浴锅水浴5min对连接酶进行灭活。然后将产物进行后续连接或置于-20℃保存。
8、连接产物转化感受态细胞
(1)感受态细胞的制备
Top 10感受态细胞的制备参照J.萨姆布鲁克(1998)《分子克隆实验指南》的方法进行。挑取Top 10单菌落接种于4mL LB培养液中,37℃,200~220r/min振荡培养12小时。取15μL菌液加到1.5mL LB液体培养基中,200r/min振荡培养3小时,此时细菌为对数生长期。冰浴10分钟,4℃4,000r/min离心10分钟,弃上清,收集菌体。加入500μL预冷的0.1M CaCl2重悬菌体。4℃,4,000r/min离心10分钟,弃上清,收集菌体,用500μL预冷的0.1M CaCl2重悬菌体。置冰上30分钟,4℃,4100r/min离心10分钟,弃上清,收集菌体,用100μL 0.1M CaCl2重悬菌体,冰浴直接转化,或加终浓度为10%的甘油,-80℃保存。
(2)连接产物转化感受态细胞
将获得的连接产物10μL加入100μL Top 10感受态细胞中,同时设立不加任何质粒的感受态细胞做阴性对照及pET-32a质粒为阳性对照。轻轻混匀,冰浴30min,42℃热击90秒后立刻冰浴10min,然后加入750μL LB液体培养基中,37℃,200r/min振摇培养1h。振荡培养后4,000r/min离心5min,弃上清,沉淀重悬于150μL LB培养液中,吹打混匀。取上述菌液涂布于含氨苄青霉素的LB琼脂平板上,待菌液完全吸收后,倒置放入37℃温箱培养12~18h,观察结果。
9、重组菌的筛选和鉴定
从转化完成的LB琼脂平板上挑取大小均一、形态圆润的疑似阳性单菌落,通过无菌操作将各个待检菌落挑至含有30μL氨苄青霉素的LB液体培养基的1.5mL的无菌离心管中,移液器吹打均匀成细菌悬浮液,同时以此悬浮液做模板进行菌落PCR鉴定(PCR体系如表8),引物对序列如表1所述。基因组全长各段基因扩增分别用对应的引物进行扩增。
表8PCR体系
PCR扩增程序:
扩增产物进行1~2%的琼脂糖凝胶电泳,选取能扩出强且特异目的条带的对应的菌液至试管氨苄青霉素LB液体培养液中摇菌,并无菌吸取300μL菌液于1.5mL Ep管中,送上海英骏生物技术有限公司进行测序。
10、全基因组测序结果处理与分析
测序结果回来后,利用Lasergene 7.10生物信息学软件中的Editseq软件进行去除包括引物对在内的外向端序列(例如图3所示),全长序列的拼接(如图4所示)及同源性分析。同时,运用NCBI中GenBank快速比对工具BLAST进行测序结果的验证或比对搜索。
实施例3上述分段扩增猪流行性腹泻病毒全基因的引物组及方法应用案例
基于本发明人研究室成功分离得到的新发流行的PEDV野毒株,为了更好更便利地研究新毒株,为此,发明人研发了本发明上述基因扩增方法。并成功运用本发明所述的引物组和方法得到了该PEDV野毒株序列的全长序列信息。
将所述PEDV野毒株全长序列经过美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)的DNA数据库,运用相似性比较的分析工具程序(Basic Local Alignment Search Tool,BLAST),与公开数据库进行相似性序列比较。
经初步研究,本发明人研究室发现的两株野毒株:CH/GDZH01/1311毒株与CH/GDZH02/1401毒株的核苷酸相似性为99.7%。而他们分别于二十世纪七十年代分离得到的CV777(AF3535111)毒株的核苷酸相似性分别仅为96.8%和96.6%。
另外,将CH/GDZH02/1401毒株传代65代次后,即为CH/GDZH02/1401-P65毒株,其相对于亲本毒株CH/GDZH02/1401发生了基因序列上多达14处氨基酸突变。不仅如此,尤其CH/GDZH02/1401-P65毒株在S基因胞质区,出现连续2个核苷酸的缺失,导致S蛋白翻译的提前终止。有研究表明,冠状病毒的此类这种现象可引起S基因过表达。此结果提示如果该传代病毒的负责刺激机体产生主要抗原的纤突蛋白S过表达,将有益于疫苗开发。
再者,CH/GDZH02/1401-P65毒株在ORF3基因出现了连续6个核苷酸的缺失,这一位置的缺失是目前NCBI数据库上的毒株都没有被公开的新现象。且这一位点的核苷酸缺失尚未有相关试验证明其功能,所以此位点的发现是一个新的发现同时也将是本发明人研究室后续研究的重点对象之一。
最后,本发明人已经将本研究室分离得到的两株PEDV野毒株和其中一株野毒株细胞传代株的基因组序列信息提交到NCBI,其中CH/GDZH01/1311毒株接收号为KR153325、CH/GDZH02/1401毒株的接收号为KR153326,而传代毒株CH/GDZH02/1401-P65毒株的接收号为KX016034。
SEQUENCE LISTING
<110> 华南农业大学
<120> 一种分段扩增猪流行性腹泻病毒全基因组的引物组及方法
<130> 2016
<160> 44
<170> PatentIn version 3.3
<210> 1
<211> 25
<212> DNA
<213> 5'F
<400> 1
acttaaagag attttctatc tacgg 25
<210> 2
<211> 19
<212> DNA
<213> 5'R
<400> 2
actggcacga tgttaccac 19
<210> 3
<211> 20
<212> DNA
<213> 1F
<400> 3
agccgtctca tactattctg 20
<210> 4
<211> 20
<212> DNA
<213> 1R
<400> 4
gaacataaga gccaaccttg 20
<210> 5
<211> 19
<212> DNA
<213> 2F
<400> 5
tgccactctt agtatcgtt 19
<210> 6
<211> 23
<212> DNA
<213> 2R
<400> 6
tcatagatgt agtacttagg cac 23
<210> 7
<211> 22
<212> DNA
<213> 3F
<400> 7
gagactatta tggctgtgct ta 22
<210> 8
<211> 18
<212> DNA
<213> 3R
<400> 8
tggcgatcac caacacat 18
<210> 9
<211> 19
<212> DNA
<213> 4F
<400> 9
attcaggtgt tgctcttac 19
<210> 10
<211> 18
<212> DNA
<213> 4R
<400> 10
accataccag tgtcttga 18
<210> 11
<211> 26
<212> DNA
<213> 5F
<400> 11
acaacattcc tagataatgg taacgg 26
<210> 12
<211> 20
<212> DNA
<213> 5R
<400> 12
tcatcaaaac ccaatgtgct 20
<210> 13
<211> 21
<212> DNA
<213> 6F
<400> 13
atagttttgg caaagacctg t 21
<210> 14
<211> 20
<212> DNA
<213> 6R
<400> 14
ggctgaaaag gcataaacca 20
<210> 15
<211> 25
<212> DNA
<213> 7F
<400> 15
gtttcttatt tacaaagttt aagcg 25
<210> 16
<211> 20
<212> DNA
<213> 7R
<400> 16
aaactcatct gtcaacgagg 20
<210> 17
<211> 21
<212> DNA
<213> 8F
<400> 17
gcttagttct tctaggattg c 21
<210> 18
<211> 20
<212> DNA
<213> 8R
<400> 18
ctattaactg ctcgtgcctc 20
<210> 19
<211> 18
<212> DNA
<213> 9F
<400> 19
ttgaccgtga ggcttcta 18
<210> 20
<211> 18
<212> DNA
<213> 9R
<400> 20
cgaccatcct tccaagtg 18
<210> 21
<211> 24
<212> DNA
<213> 10F
<400> 21
aaaagatgta ccaagtctgc gatg 24
<210> 22
<211> 22
<212> DNA
<213> 10R
<400> 22
tagtaccaat aacaaccgaa gc 22
<210> 23
<211> 20
<212> DNA
<213> 11F
<400> 23
gtgtttcgct cttgtcaacc 20
<210> 24
<211> 19
<212> DNA
<213> 11R
<400> 24
cgctgcaaac agacgcaat 19
<210> 25
<211> 21
<212> DNA
<213> 12F
<400> 25
ttaatcgcat tgctacatcc g 21
<210> 26
<211> 20
<212> DNA
<213> 12R
<400> 26
ccacaaccct cattagcctg 20
<210> 27
<211> 20
<212> DNA
<213> 13F
<400> 27
tataatgtgc gataggtccc 20
<210> 28
<211> 20
<212> DNA
<213> 13R
<400> 28
tgccacagat tataggtgtc 20
<210> 29
<211> 21
<212> DNA
<213> 14F
<400> 29
cttagggcta gtaactgcat t 21
<210> 30
<211> 22
<212> DNA
<213> 14R
<400> 30
tataatcagc atcgctaacg ta 22
<210> 31
<211> 20
<212> DNA
<213> 15F
<400> 31
aaaatgctgt gcttatgtca 20
<210> 32
<211> 18
<212> DNA
<213> 15R
<400> 32
tgtggtaggc taagtgtt 18
<210> 33
<211> 25
<212> DNA
<213> 16F
<400> 33
cattagtgat gttgtgttag gcttg 25
<210> 34
<211> 22
<212> DNA
<213> 16R
<400> 34
gaaggtttct agaagagata gg 22
<210> 35
<211> 22
<212> DNA
<213> 17F
<400> 35
cgactaattt tgttgatgca ct 22
<210> 36
<211> 22
<212> DNA
<213> 17R
<400> 36
tgcagcagta aaacctccta gc 22
<210> 37
<211> 20
<212> DNA
<213> 18F
<400> 37
tcacatgtat agtgcgtctc 20
<210> 38
<211> 23
<212> DNA
<213> 18R
<400> 38
gctgccaaca taatataatt gcg 23
<210> 39
<211> 20
<212> DNA
<213> 19F
<400> 39
atctacttct ttgcactgtt 20
<210> 40
<211> 20
<212> DNA
<213> 19R
<400> 40
atttgctggt ccttatttcc 20
<210> 41
<211> 21
<212> DNA
<213> 20F
<400> 41
attcagctgt gagtaatccg a 21
<210> 42
<211> 19
<212> DNA
<213> 20R
<400> 42
accgttgtgt gcaagacca 19
<210> 43
<211> 28
<212> DNA
<213> 3'F
<400> 43
gggtgcgcca tctgatgtga cccatgcc 28
<210> 44
<211> 20
<212> DNA
<213> 3'R
<400> 44
gtgtatccat atcaacaccg 20
- 还没有人留言评论。精彩留言会获得点赞!