取代的氨基吡喃衍生物的晶型的制作方法
本发明涉及一种取代的氨基吡喃衍生物,或其水合物、溶剂化物的晶型,以及其制备方法或其药物组合物和其在制备二肽激酶-iv(dpp-iv)抑制剂药物上的用途。
背景技术:
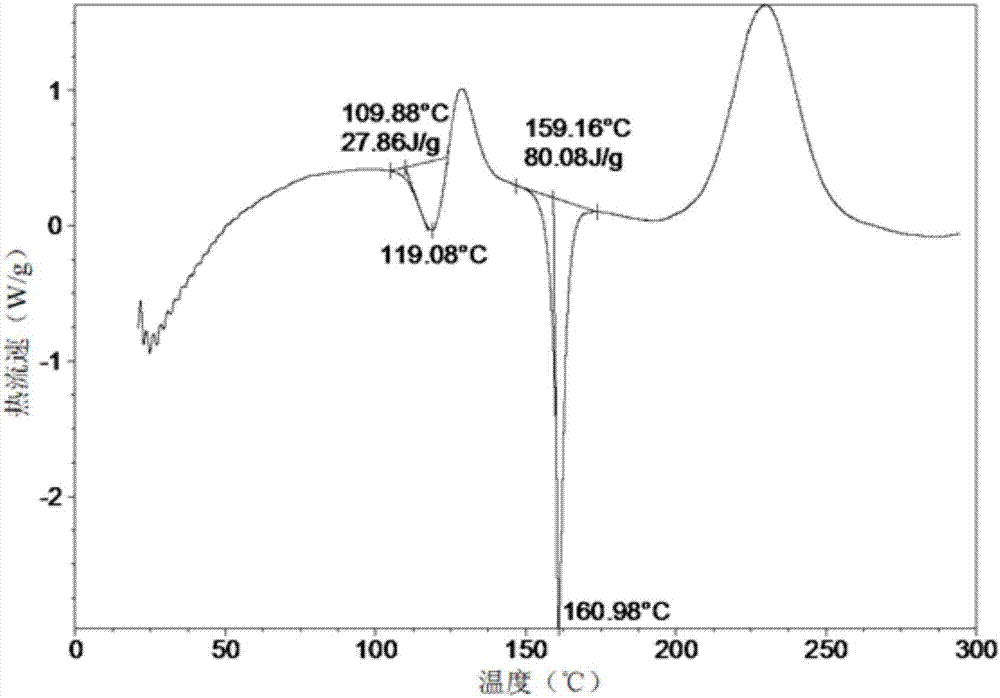
:二肽基肽酶-iv(dipeptidylpeptidase,dpp-iv,ec3.4.14.5)是一个丝氨酸蛋白酶,从含有l-脯氨酸和l-丙氨酸的多肽n端倒数第二位水解n端二肽。通过增强肠促胰岛素活性发挥作用,属于非胰岛素治疗药物。dpp-iv抑制剂无体重增加和水肿等不良反应。pct/cn2015/078923专利申请公开了新的吡喃衍生物(2r,3s,5r,6s)-2-(2,5-二氟苯基)-5-(2-(甲基磺酰基)-吡咯并[3,4]吡唑-5(2h,4h,6h)-基)-6-(三氟甲基)四氢-2h-吡喃-3-胺,结构式如式(i),本文简称为化合物a。该结构显示对dpp-iv具有良好的抑制作用,具有用于预防和/或治疗ii型糖尿病的潜能。技术实现要素:本发明提供了(2r,3s,5r,6s)-2-(2,5-二氟苯基)-5-(2-(甲基磺酰基)-吡咯并[3,4]吡唑-5(2h,4h,6h)-基)-6-(三氟甲基)四氢-2h-吡喃-3-胺,即化合物a的晶型及其化合物a的水合物的晶型、溶剂化物的晶型,化合物a具有以下化学结构(i):本发明新晶型具有优点,诸如易于加工和结晶、处理、溶解度好、稳定性强、压力稳定性和给药,这些使它们尤其适于各种药物剂型的制造。本发明的晶型表现出优于化合物a的无定形游离碱的制药学优势。尤其是,晶型增强了化学和物理的稳定性,更有利于在制备包含药理学活性成分的固体药物剂型。本发明的晶型以原料药的约5重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约10重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约15重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约20重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约25重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约30重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约35重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约40重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约45重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约50重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约55重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约60重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约65重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约70重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约75重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约80重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约85重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约90重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约95重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约98重量%至约100重量%存在。在某些实施方案中,本发明的晶型以原料药的约99重量%至约100重量%存在。在某些实施方案中,基本上所有的原料药都是本发明的晶型,即原料药基本上是相纯晶体。本发明化合物a无特殊说明,则为化合物a的无定形态。本发明所述晶型的一个实施方案为无水化合物a(晶型i),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:7.6°±0.2°、9.2°±0.2°、13.3°±0.2°、16.1°±0.2°、17.1°±0.2°和18.7°±0.2°。其中该晶型i的x-射线粉末衍射中,2θ衍射角度还在10.5°±0.2°、15.0°±0.2°、17.8°±0.2°、20.6°±0.2°、23.4°±0.2°和24.2°±0.2°处具有特征衍射峰。进一步的,该晶型i的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:21.4°±0.2°、22.6°±0.2°和26.4°±0.2°。再进一步,晶型i的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:10.7°±0.2°、12.2°±0.2°、12.8°±0.2°、14.4°±0.2°、24.6°±0.2°、25.9°±0.2°、28.2°±0.2°、29.2°±0.2°、32.2°±0.2°和34.1°±0.2°。更进一步,该晶型i的x-射线粉末衍射图谱(xrd)基本如附图1所示。本发明所述的晶型i,其差示扫描量热分析曲线(dsc)显示一条吸热曲线,其中t开始=109.88℃,t峰=119.08℃,△h=27.86j/g,熔融伴随转晶,转晶后样品的t开始=159.16℃,t峰=160.98℃,△h=80.08j/g。本发明所述的晶型i,其差示扫描量热分析曲线(dsc)如附图2所示。本发明所述的晶型i,其热重分析曲线(tga)显示在150℃之前有一段约1.6%(w/w)的缓慢失重(因为样品中具有残余溶剂),分解温度为219.8℃。本发明所述的晶型i,其热重分析曲线(tga)如附图3所示。本发明所述晶型的一个实施方案为无水化合物a(晶型ii),晶型ii使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:8.2°±0.2°、14.9°±0.2°、16.5°±0.2°、17.4°±0.2°、18.7°±0.2°和20.3°±0.2°。其中,该晶型ii的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:12.0°±0.2°、13.1°±0.2°、14.0°±0.2°、21.3°±0.2°、21.9°±0.2°和23.9°±0.2°。进一步地,该晶型ii的x-射线粉末衍射图谱还在10.0°±0.2°、10.9°±0.2°、24.9°±0.2°、26.5°±0.2°、26.9°±0.2°和28.4°±0.2°的2θ位置具有特征衍射峰。再进一步地,该晶型ii的x-射线粉末衍射图谱还在15.8°±0.2°、18.0°±0.2°、23.0°±0.2°、25.7°±0.2°和27.9°±0.2°。更进一步,该晶型ii的x-射线粉末衍射图谱基本如附图4所示。本发明所述的晶型ii,其差示扫描量热分析曲线(dsc)显示晶型ii的t开始=197.97℃,t峰=202.10℃,△h=75.51j/g。本发明所述的晶型ii,其差示扫描量热分析曲线(dsc)如附图5所示。本发明所述的晶型ii,其热重分析曲线(tga)显示样品在150℃之前失重约0.7%,分解温度为220.5℃。本发明所述的晶型ii,其热重分析曲线如图6所示。本发明所述化合物a的晶体可以是二水合物。其中二水合物化合物a(晶型iii)使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:6.8°±0.2°、8.4°±0.2°、16.7°±0.2°、17.8°±0.2°、19.1°±0.2°和23.8°±0.2°。其中,该晶型iii的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:6.0°±0.2°、9.8°±0.2°、14.4°±0.2°、15.0°±0.2°、19.9°±0.2°和21.6°±0.2°。进一步,晶型iii的x-射线粉末衍射图谱还在10.4°±0.2°、10.8°±0.2°、11.3°±0.2°、11.9°±0.2°、12.4°±0.2°和16.3°±0.2°的2θ位置具有特征衍射峰。再进一步,晶型iii的x-射线粉末衍射图谱还在13.7°±0.2°、20.9°±0.2°、22.0°±0.2°、22.8°±0.2°、24.3°±0.2°、25.5°±0.2°、28.0°±0.2°、29.9°±0.2°和31.2°±0.2°位置具有特征衍射峰。更进一步,晶型iii的x-射线粉末衍射图谱基本如附图7所示。本发明所述的晶型iii,其差示扫描量热分析曲线(dsc)显示一条吸热曲线,其中t开始=97.22℃,t峰=109.52℃,△h=53.87j/g,脱水后转晶,转晶后样品的t开始=160.91℃,t峰=163.05℃,△h=27.79j/g。本发明所述的晶型iii,其差示扫描量热分析曲线如附图8所示。本发明所述的晶型iii,其热重分析曲线(tga)显示在170℃之前失重约7.3%,约合含两分子水(7.2%),分解温度为215.7℃;其热重分析曲线如图9所示。本发明的一个实施方案化合物a的甲苯溶剂化物(晶型v),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:5.6°±0.2°、12.0°±0.2°、14.9°±0.2°、17.3°±0.2°、19.3°±0.2°和22.5°±0.2°。其中,晶型v的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:9.4°±0.2°、12.3°±0.2°、12.9°±0.2°、16.7°±0.2°、19.8°±0.2°和22.9°±0.2°。进一步地,晶型v的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:8.5°±0.2°、16.1°±0.2°、17.7°±0.2°、21.1°±0.2°、23.5°±0.2°、24.2°±0.2°和24.6°±0.2°。更进一步,晶型v的x-射线粉末衍射图谱基本如附图10所示。本发明所述的晶型v,其差示扫描量热分析曲线(dsc)显示一条吸热曲线,其中t开始=76.13℃,t峰=88.26℃,△h=72.23j/g,转晶后样品t开始=158.46℃,t峰=160.99℃,△h=73.44j/g。本发明所述的晶型v,其差示扫描量热分析曲线如附图11所示。本发明所述的晶型v,其热重分析曲线(tga)显示在100℃之前失重10.0%,约合含半分子甲苯(9.0%),分解温度为221.1℃。本发明所述的晶型v,热重分析曲线如附图12所示。本发明的一个实施方案化合物a(晶型vi),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:5.3°±0.2°、7.9°±0.2°、13.3°±0.2°、17.4°±0.2°、17.9°±0.2°和19.2°±0.2°。其中,晶型vi的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:7.5°±0.2°、15.5°±0.2°、16.0°±0.2°、19.7°±0.2°、20.1°±0.2°和24.2°±0.2°。进一步,晶型vi的x-射线粉末衍射图谱基本如附图13所示。本发明的一个实施方案化合物a(晶型vii),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:3.8°±0.2°、7.0°±0.2°、8.9°±0.2°、14.5°±0.2°、17.3°±0.2°、18.2°±0.2°和19.8°±0.2°。其中,晶型vii的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:7.4°±0.2°、10.0°±0.2°、11.8°±0.2°、13.5°±0.2°、14.8°±0.2°、16.1°±0.2°和20.4°±0.2°。进一步,晶型vii的x-射线粉末衍射图谱基本如附图14所示。本发明的一个实施方案化合物a(晶型viii),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:6.9°±0.2°、8.6°±0.2°、15.7°±0.2°、17.4°±0.2°、18.1°±0.2°和21.3°±0.2°。其中,晶型viii的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:11.6°±0.2°、18.7°±0.2°、23.5°±0.2°、26.2°±0.2°和28.7°±0.2°。进一步,晶型viii的x-射线粉末衍射图谱基本如附图15所示。本发明的一个实施方案化合物a(晶型ix),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:7.8°±0.2°、9.6°±0.2°、15.0°±0.2°、17.8°±0.2°、21.2°±0.2°和25.7°±0.2°。其中,晶型ix的x-射线粉末衍射图谱基本如附图16所示。本发明的一个实施方案化合物a(晶型x),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:6.0°±0.2°、6.7°±0.2°、7.7°±0.2°、13.9°±0.2°、17.4°±0.2°和19.2°±0.2°。其中,晶型x的x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:9.6°±0.2°、11.9°±0.2°、14.4°±0.2°、18.5°±0.2°、21.5°±0.2°、23.5°±0.2°和23.9°±0.2°。进一步,晶型x的x-射线粉末衍射图谱基本如附图17所示。本发明的一个实施方案化合物a(晶型xi),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:5.7°±0.2°、11.4°±0.2°、16.7°±0.2°、18.0°±0.2°、19.0°±0.2°、19.5°±0.2°和21.5°±0.2°。其中,晶型xi的x-射线粉末衍射图谱基本如附图18所示。本发明的一个实施方案化合物a(晶型xii),使用cu-kα辐射,其x-射线粉末衍射图谱在以下2θ位置具有特征衍射峰:6.7°±0.2°、7.4°±0.2°、8.9°±0.2°、13.6°±0.2°、16.3°±0.2°、18.0°±0.2°和23.5°±0.2°。其中,晶型xii的x-射线粉末衍射图谱还在以下2θ位置具有特征衍射峰:9.5°±0.2°、15.4°±0.2°、17.1°±0.2°、20.7°±0.2°、22.8°±0.2°和24.6°±0.2°。进一步,晶型xii的x-射线粉末衍射图谱基本如附图19所示。可以理解的是,差示扫描量热(dsc)领域中所熟知的,dsc曲线的熔融峰高取决于与样品制备和仪器几何形状有关的许多因素,而峰位置对实验细节相对不敏感。因此,在一些实施方案中,本发明的结晶化合物具有特征峰位置的dsc图,具有与本发明附图中提供的dsc图实质上相同的性质,误差容限为±3℃。本发明还涉及一种药物组合物,包含治疗有效量的本发明中所述的晶型化合物,以及一种或多种药学上可接受的载体或赋形剂。本发明所述的晶型作为活性药物成分,或其作为活性成分的药物组合物可以用于制备预防和/或治疗糖尿病、糖尿病性视网膜病、糖尿病性神经病、糖尿病性肾病的药物,优选用于制备ii型糖尿病药物。本发明还公开了一种治疗代谢性疾病的方法,该方法包括给药本发明所述的一种或多种晶型化合物a,或其药物组合物。本发明公开的x-射线粉末衍射或dsc图、tga图,与其实质上相同的也属于本发明的范围。除非有相反的陈述,在说明书和权利要求书中使用的术语具有下述含义。“有效剂量”指引起组织、系统或受试者生理或医学翻译的化合物的量,此量是所寻求的,包括在受治疗者身上施用时足以预防受治疗的疾患或病症的一种或几种症状发生或使其减轻至某种程度的化合物的量。“ic50”指半数抑制浓度,指达到最大抑制效果一半时的浓度。本发明晶型结构可以使用本领域普通技术人员已知的各种分析技术分析,包括但不限于,x-射线粉末衍射(xrd)、示差扫描热法(dsc)和/或热重分析(thermogravimetricanalysis,tga)。热重分析(thermogravimetricanalysis,tga),又叫热重法(thermogravimetry,tg)。本发明使用的x-射线粉末衍射仪(xrd)为brukerd8advancediffractometer,铜靶波长为1.54nm的kαradiation(40kv,40ma),θ-2θ测角仪,mo单色仪,lynxeye探测器,校准物质al2o3,采集软件为diffracplusxrdcommander,分析软件为mdijade6;方法参数:无反射样品板规格为24.6mmdiameterx1.0mmthickness,无反射样品板厂家为mticorporation,变温热台厂家为上海微图仪器科技发展有限公司,变温热台样品板为铜板,检测角度:3-40°2θ,步长:0.02°2θ。本发明使用的差热分析扫描仪(dsc)为tainstrumentsq200dsc,氮气保护,气体流速为40ml/分钟。本发明使用的热重分析仪(tga)为tainstrumentsq500tga,氮气保护,气体流速为40ml/分钟。可以理解的是,本发明描述的和保护的数值为近似值。数值内的变化可能归因于设备的校准、设备误差、晶体的纯度、晶体大小、样本大小以及其他因素。可以理解的是,本发明的晶型不限于与本发明公开的附图中描述的特征图谱完全相同的特征图谱,比如xrd、dsc、tga,具有与附图中描述的哪些图谱基本上相同或本质上相同的特征图谱的任何晶型均落入本发明的范围内。本发明公开的晶型可以经如下的常见的制备晶型的方法制备:1、挥发实验是将样品澄清溶液在不同温度下敞口挥发至溶剂干。2、晶浆实验是将样品的过饱和溶液(有不溶固体存在)在不同溶剂体系中某个温度下进行搅拌。3、抗溶剂实验是取样品溶解在良溶剂中,加入抗溶剂,析出固体短时搅拌后立即过滤处理。4、冷却结晶实验是在高温下将一定量的样品溶解到相应溶剂中,然后直接在室温或低温搅拌析晶。5、高分子模板实验是在样品澄清溶液中加入不同种类的高分子材料,置于室温下敞口挥发至溶剂干。6、热方法实验是将样品按一定热方法结晶条件处理并冷却至室温。7、水汽扩散实验是将样品在室温下一定湿度环境中放置。在不背离本发明的范围和主旨下,通过考虑本发明的说明书和实施例操作内容后,对本发明进行各种不同的改进和改变对本领域技术人员将是显而易见的。附图说明图1是化合物a晶型i使用cu-kα辐射的x-射线粉末衍射图谱。图2是化合物a晶型i的差示扫描量热法(dsc)曲线。图3是化合物a晶型i的热重分析(tga)曲线。图4是化合物a晶型ii使用cu-kα辐射的x-射线粉末衍射图谱。图5是化合物a晶型ii的差示扫描量热法(dsc)曲线。图6是化合物a晶型ii的热重分析(tga)曲线。图7是化合物a晶型iii使用cu-kα辐射的x-射线粉末衍射图谱。图8是化合物a晶型iii的差示扫描量热法(dsc)曲线。图9是化合物a晶型iii的热重分析(tga)曲线。图10是化合物a晶型v使用cu-kα辐射的x-射线粉末衍射图谱。图11是化合物a晶型v的差示扫描量热法(dsc)曲线。图12是化合物a晶型v的热重分析(tga)曲线。图13是化合物a晶型vi使用cu-kα辐射的x-射线粉末衍射图谱。图14是化合物a晶型vii使用cu-kα辐射的x-射线粉末衍射图谱。图15是化合物a晶型viii使用cu-kα辐射的x-射线粉末衍射图谱。图16是化合物a晶型ix使用cu-kα辐射的x-射线粉末衍射图谱。图17是化合物a晶型x使用cu-kα辐射的x-射线粉末衍射图谱。图18是化合物a晶型xi使用cu-kα辐射的x-射线粉末衍射图谱。图19是化合物a晶型xii使用cu-kα辐射的x-射线粉末衍射图谱。具体实施方式以下通过具体实施例详细说明本发明的实施过程和产生的有益效果,旨在帮助阅读者更好地理解本发明的实质和特点,不作为对本案可实施范围的限定。实施例中无特殊说明,溶液是指水溶液。除非特殊说明,结晶的实验条件一般为室温(20-30℃,30-70%rh),溶剂比例是指体积比。实施例1化合物a的制备:第一步至第三步参考专利wo2015/192701制备。第四步:(2r,3s,5r,6s)-2-(2,5-二氟苯基)-5-(2-(甲基磺酰基)-吡咯并[3,4]吡唑-5(2h,4h,6h)-基)-6-(三氟甲基)四氢-2h-吡喃-3-胺(化合物a)(2r,3s,5r,6s)-2-(2,5-difluorophenyl)-5-(2-(methylsulfonyl)pyrrolo[3,4-c]pyrazol-5(2h,4h,6h)-yl)-6-(trifluoromethyl)tetrahydro-2h-pyran-3-amine0℃下氮气氛围,将1c(57.5g,101.6mmol)溶于二氯甲烷(345ml)和三氟乙酸(86ml)中,加毕,室温下搅拌4小时。反应结束,在低于20℃下,将反应液减压浓缩至黏稠,加入水(600ml)和二氯甲烷(80ml),搅拌静置,分层,水相中加入二氯甲烷(300ml),搅拌状态下用氨水调节ph值至9-10,分层,用二氯甲烷(300ml×2)萃取水相。合并有机相,有机相用水(300ml)洗涤,无水硫酸钠干燥,浓缩。硅胶柱层析分离纯化(乙酸乙酯/甲醇(v/v)=50:1),干燥,旋干,得到白色粉末固体化合物a(37.8g,产率80%)。使用xrd、dsc和tga分析,为化合物a晶型i,见图1-3。实施例2化合物a晶型ii的制备方法一:50℃下,将化合物a晶型i(50mg)溶于水(5.0ml)和丙酮(2.8ml)中,热过滤,滤液在3℃下搅拌2天,过滤,滤饼在室温下真空干燥得化合物a晶型ii。通过xrd、dsc和tga表征化合物a晶型ii,见图4-6。方法二:化合物a晶型i(10mg)加入异丙醚(1.0ml)与丙酮(0.1ml)的混合溶剂中,在室温下晶浆3天。取晶浆后的悬浊液离心即可得到化合物a晶型ii,谱图与图4-6相同。实施例3化合物a晶型iii的制备方法一:室温下,将化合物a晶型i(50mg)溶于四氢呋喃(0.5ml)中,过滤,搅拌条件下滴加甲基环己烷(5.0ml),析出大量白色固体,继续搅拌10分钟后过滤,将滤饼在室温下真空干燥得化合物a晶型iii。通过xrd、dsc和tga表征化合物a晶型iii,见图7-9。方法二:将化合物a晶型i(10mg)溶于水/四氢呋喃(0.5ml/1.0ml)、水/乙醇(0.5ml/2ml)、正庚烷/甲醇(0.5ml/0.5ml)或水饱和的氯仿溶液(0.5ml),放置在室温下自然挥发至干,或者将化合物a晶型i(10mg)溶于水/乙腈(0.5ml/0.5ml),40℃下自然挥发干,得到化合物a晶型iii,谱图与图7-9相同。方法三:将化合物a晶型i(10mg)放置于小瓶中,加入异丙醇(3ml)或者甲基叔丁基醚(2ml),得到溶清液,放置在室温下自然挥发干,得到化合物a晶型iii,谱图与图7-9相同。方法四:将化合物a晶型i(10mg)放置于小瓶中,加入水/1,4-二氧六环(1.0ml/0.5ml)、水/乙腈(1.0ml/0.2ml)或者乙醇/甲苯(0.2ml/0.1ml)溶剂,得到悬浊液,在40℃下晶浆3天。取晶浆后的悬浊液离心,得到化合物a晶型iii,谱图与图7-9相同。实施例4化合物a晶型v的制备室温下,化合物a晶型i(50mg)中加入甲苯(4.0ml),超声15分钟,体系为凝胶状态,直接置于室温,敞口自然挥发至干得到化合物a晶型v。通过xrd、dsc和tga表征化合物a晶型v,见图10-12。实施例5化合物a晶型vi的制备室温下,将化合物a晶型i(50mg)溶于二氯甲烷(1.0ml)中,过滤,室温旋干得化合物a晶型vi。通过xrd表征化合物a晶型vi,见图13。实施例6化合物a晶型vii的制备升温将化合物a晶型i(50mg)溶于乙醚(3.0ml)中,过滤,室温旋干得化合物a晶型vii。通过xrd表征化合物a晶型vii,见图14。实施例7化合物a晶型viii的制备50℃下,将化合物a晶型i(10mg)溶于水(1.0ml)和丙酮(0.6ml)中,热过滤,直接放在3℃条件下搅拌析晶,离心,得到化合物a晶型viii。通过xrd表征化合物a晶型viii,见图15。实施例8化合物a晶型ix的制备70℃下,将化合物a晶型i(10mg)溶于乙醇(0.4ml)中,热过滤,室温搅拌析晶,得到化合物a晶型ix。通过xrd表征化合物a晶型ix,见图16。实施例9化合物a晶型x的制备方法一:室温下,将化合物a晶型i(10mg)溶于乙醇(2.0ml)中,过滤,滤液放置于室温下自然挥发至干,得到化合物a晶型x。通过xrd表征化合物a晶型x,见图17。方法二:化合物a晶型i(10mg)溶于甲醇(0.4ml)或乙醇(2ml)中,室温下自然挥发至干,得到化合物a晶型x,谱图与图17相同。方法三:化合物a晶型i(10mg)溶于乙醇/异丙醚(2ml/0.5ml)、乙醇/二氯甲烷(0.5ml/0.5ml)或者乙醇/正庚烷(2.0ml/0.5ml)中,室温下自然挥发至干,得到化合物a晶型x,谱图与图17相同。实施例10化合物a晶型xi的制备50℃下,将化合物a晶型i(10mg)溶于甲基叔丁基醚(0.7ml)中,热过滤,3℃条件下搅拌析晶,析出固体后立刻离心并直接取固体样得到化合物a晶型xi。通过xrd表征化合物a晶型xi,见图18。实施例11化合物a晶型xii的制备方法一:70℃下,将化合物a晶型i(10mg)溶于甲基环己烷(1.0ml)和乙醇(0.2ml),热过滤,滤液在室温搅拌析晶,析出固体得到化合物a晶型xii。通过xrd表征化合物a晶型xii,见图19。方法二:化合物a晶型i(10mg)溶于乙醇/甲基叔丁基醚(1.0ml/1.0ml),放置在40℃下自然挥发至干,得到化合物a晶型xii,谱图与图19相同。实施例12晶型稳定性及转化1.化合物a晶型i~xii在不同的析晶环境或使用不同的析晶方法,可以转换为化合物a晶型ii或化合物a晶型iii。见表1。表1化合物a晶型i~xii转化关系转换前晶型转换条件转换后晶型晶型viii水/丙酮中搅拌2天晶型ii晶型ii室温干燥放置14天晶型ii晶型iii室温干燥放置14天晶型iii参照本文所述的制备晶型xii的方法,放大反应量至50mg时,得到的为晶型iii。化合物a晶型ii、iii稳定性较好,其他晶型在不同的条件下可以转化为晶型ii或iii。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 含磷铝结构单元的mcm-41分子筛催化剂、制备方法及其应用的制作方法
- 三维有序大孔结构Ag微米束/Eu<sub>0.6</sub>Sr<sub>0.4</sub>FeO<sub>3</sub>复合催化剂、制备及应用的制作方法
- 三维有序大孔结构CoO<sub>x</sub>/Eu<sub>0.6</sub>Sr<sub>0.4</sub>FeO<sub>3</sub>催化剂、制备方法及应用的制作方法
- 一种铁同晶取代分子筛的方法
- 制备同晶取代硅酸盐的方法
- 高硅沸石及其杂原子同晶取代衍生物的合成的制作方法
- 一种低杂质含量铜基甲醇合成催化剂的制备方法
- 层柱粘土催化剂的制备方法
- 一种通过固相同晶取代制备杂原子沸石的方法
- 一种无机微孔磷酸镍钴类分子筛材料及其合成方法