一类含吡唑杂环的熊果酰胺衍生物及其合成与应用的制作方法
本发明属于化学制药技术领域,涉及一类含吡唑杂环的熊果酰胺衍生物及其合成与应用,具体涉及一类含吡唑杂环的熊果酰胺衍生物及其合成方法且在治疗肿瘤的药物的应用。
背景技术:
现代医学证明,肿瘤是机体在环境污染、化学污染、电离辐射、自由基毒素、微生物(细菌、真菌、病毒等)及其代谢毒素、遗传特性、内分泌失衡、免疫功能紊乱等各种生理生化因素作用下,导致局部组织的某些细胞在基因水平上失去对其生长的正常调控,克隆性异常增殖而形成的新生物。临床上,肿瘤治疗主要采取手术、放射、化疗、免疫及中医药等多种措施。化疗即化学药物治疗,属于肿瘤的全身治疗方法,目前已有80余种化疗药物应用于临床,多种恶性肿瘤的化疗治愈率超过50%。现阶段临床使用的抗肿瘤药物,可分为烷化剂、铂化合物、抗生素、天然药物及激素五大类。其中,约有1/3的药物来自于植物。从天然植物中分离得到的抗肿瘤活性成分,代表性药物有紫杉醇、长春新碱、替尼泊苷等。天然抗肿瘤药物的抗癌谱广,选择性高,毒副作用小,在临床应用上有着十分广阔的发展前景。目前,从天然植物中提取、分离抗肿瘤活性成分、开发毒副作用小、专一性高、杀伤力强的天然抗肿瘤药物,已成为近年来国内外抗肿瘤药物研究的热点。
熊果酸(ursolicacid,ua),是一种存在于天然植物中的乌苏烷型五环三萜类化合物,具有镇静、抗炎、护肝、降血脂等多种生物活性,是已知多种中草药的主要活性成分。近年来,国内外陆续报道了熊果酸对多种肿瘤细胞增殖的显著抑制作用,具有确定的诱导肿瘤细胞凋亡的作用。然而由于熊果酸类化合物较差的溶解性、靶点不明确以及生物利用度较低,限制了其在药物开发中的发展。近年来,随着人们对天然产物及其类似物的成药性研究再度兴起,通过化学手段对熊果酸的结构修饰以降低副作用、提高生物活性及改善其生物利用度,再次成为萜类天然产物药物开发的热点。
根据2014年康奈尔大学njardarson教授对fda批准上市药物的结构统计,发现59%药物分子均含有氮原子杂环结构。因此,基于复杂天然产物结构引入氮原子杂环结构是对其进行结构修饰的重要策略。其中,吡唑杂环是活性药物分子的重要结构骨架,被广泛的应用于天然产物的结构改造及活性药物分子的设计合成中。因此,有必要对熊果酸进行进一步的研究与探讨。
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一类含吡唑杂环的熊果酰胺衍生物及其合成与应用,用于对肿瘤细胞的生长进行抑制,具有新型抗肿瘤药物的开发潜力。
为实现上述目的及其他相关目的,本发明第一方面提供一类含吡唑杂环的熊果酰胺衍生物,所述衍生物具有如下式8所示的结构通式:
式中,
r1选自氢(h-)、c1-c4烷基、c3-c6环烷基、芳基、芳基烷基;
r2选自氢(h-)、c1-c4烷基、卤代烷基、c(o)or’;所述r’为c1-c4烷基;
r3选自氢(h-)、c1-c6烷基、c3-c6环烷基、卤代烷基、环烷基烷基、烷氧基烷基、任选取代的芳基烷基、杂环基烷基;
r4为c1-c6烷基、c3-c6环烷基、卤代烷基、环烷基烷基、烷氧基烷基、任选取代的芳基烷基、杂环基烷基;
或者,所述r3和r4与桥接的氮原子一起形成取代或未取代的杂环烷基。
优选地,所述r1中c1-c4烷基为甲基(ch3-)或异丙基(ipr-)。
优选地,所述r1中c3-c6环烷基为环戊基(cyclopentyl)。
优选地,所述r1中芳基烷基为苄基(bn-)。
优选地,所述r1中芳基为苯基(ph-)、4-溴苯基(4-brc6h5-)、3-氟苯基(3-fc6h5-)、4-氰基苯基(4-cnc6h5-)或4-羧基苯基(4-co2hc6h5-)。
优选地,所述r2中c1-c4烷基为甲基(ch3-)。
优选地,所述r2中卤代烷基为三氟甲基(cf3-)。
优选地,所述r2中c(o)or’为co2et-。
优选地,所述r3中c1-c6烷基为异丙基(isopropyl-)、正己基(n-hexyl-)或正丁基(n-butyl-)。
优选地,所述r3中c3-c6环烷基为环己基(cyclopentyl-)或环丁基(cyclobutyl-)。
优选地,所述r3中环烷基烷基为环丙基甲基(cyclopropylmethyl-)。
优选地,所述r3中卤代烷基为2,2,2-三氟乙基(2,2,2-trifluoroethyl-,
优选地,所述r3中烷氧基烷基为3-异丙氧基丙基(3-isopropoxypropyl-,
优选地,所述r3中取代的芳基烷基为4-氟苯甲基((4-fluorophenyl)methyl-,
优选地,所述r3中杂环基烷基为2-吡咯乙基(2-(pyrrolidin-1-yl)ethyl-,
优选地,所述r4中c1-c6烷基为异丙基(isopropyl-)、正己基(n-hexyl-)或正丁基(n-butyl-)。
优选地,所述r4中c3-c6环烷基为环戊基(cyclopentyl-)、环己基(cyclopentyl-)或环丁基(cyclobutyl-)。
优选地,所述r4中环烷基烷基为环丙基甲基(cyclopropylmethyl-)。
优选地,所述r4中卤代烷基为2,2,2-三氟乙基(2,2,2-trifluoroethyl-)。
优选地,所述r4中烷氧基烷基为3-异丙氧基丙基(3-isopropoxypropyl-)。
优选地,所述r4中取代的芳基烷基为4-氟苯甲基((4-fluorophenyl)methyl-)或4-甲氧基苯乙基((4-methoxyphenyl)ethyl-)。
优选地,所述r4中杂环基烷基为2-吡咯乙基(2-(pyrrolidin-1-yl)ethyl-)、4-吡啶甲基(4-pyridylmethyl-)、2-呋喃甲基(2-furanylmethyl-)或3-吗啉基丙基(3-morpholinopropyl-)。
优选地,所述r3和r4与桥接的氮原子一起形成的杂环烷基选自
优选地,所述一类含吡唑杂环的熊果酰胺衍生物为化合物lb-1、化合物lb-2、化合物lb-3、化合物lb-4、化合物lb-5、化合物lb-6、化合物lb-7、化合物lb-8、化合物lb-9、化合物lb-10、化合物lb-11、化合物lb-12、化合物lb-13、化合物lb-14或化合物lb-15,其中,所述化合物的r1、r2、r3、r4基团见下表1。
表1.化合物的r1、r2、r3、r4基团列表
本发明第二方面提供一类含吡唑杂环的熊果酰胺衍生物的合成方法,其合成路线如下:
具体包括以下步骤:
a)以熊果酸(1)为原料,使熊果酸(1)的c-28位羧基获得苄基保护,获得中间体(2);
优选地,在步骤a)中,所述熊果酸(1)为商品化的熊果酸。
优选地,在步骤a)中,所述苄基保护是将熊果酸(1)与碳酸钾(k2co3)、n,n-二甲基酰胺(dmf)、苄溴(bnbr)混合后进行加热反应,获得的混合物冷却至室温后,加水析出固体产物,将固体产物过滤、洗涤、干燥后,获得中间体(2)。
更优选地,所述熊果酸(1)与碳酸钾加入的摩尔(mol)比为1:1-3。进一步优选地,所述熊果酸(1)与碳酸钾加入的摩尔(mol)比为1:1.5。
更优选地,所述熊果酸(1)与n,n-二甲基酰胺加入的摩尔(mol)比为1:9-11。进一步优选地,所述熊果酸(1)与n,n-二甲基酰胺加入的摩尔(mol)比为1:10。
更优选地,所述熊果酸(1)与苄溴加入的摩尔(mol)比为1:1-2。进一步优选地,所述熊果酸(1)与苄溴加入的摩尔(mol)比为1:1.5。
更优选地,所述加热反应的条件为:反应温度为50-70℃;反应时间为3-5h。进一步优选地,所述加热反应的条件为:反应温度为60℃;反应时间为4h。
更优选地,所述室温为20-25℃。
更优选地,所述熊果酸(1)加入的质量(mg)与水加入的体积(ml)之比为450-470:40-60。进一步优选地,所述熊果酸(1)加入的质量(mg)与水加入的体积(ml)之比为460:50。
优选地,在步骤a)中,所述中间体(2)的产率为90-94%。
更优选地,在步骤a)中,所述洗涤采用水进行多次洗涤。
b)在中间体(2)中加入pcc进行氧化反应,使中间体(2)的c-3位羟基氧化形成羰基,获得中间体(3);
优选地,在步骤b)中,所述氧化反应是将中间体(2)溶于二氯甲烷后冷却至0℃以下,加入pcc在室温下搅拌进行氧化反应,将获得的反应产物过滤、浓缩、分离纯化后,获得中间体(3)。
更优选地,所述中间体(2)加入的质量(mg)与二氯甲烷加入的体积(ml)之比为440-460:40-60。进一步优选地,所述中间体(2)加入的质量(mg)与二氯甲烷加入的体积(ml)之比为450:50。
更优选地,所述中间体(2)与pcc加入的摩尔比为1:1.2-3。进一步优选地,所述中间体(2)与pcc加入的摩尔比为1:1.5。所述pcc为吡啶和cro3在盐酸溶液中的络合盐。
更优选地,所述搅拌时间为11-13h。进一步优选地,所述搅拌时间为12h。
优选地,在步骤b)中,所述中间体(3)的产率为83-85%。
c)将中间体(3)在碱性条件下加入酯类化合物进行反应,使中间体(3)的c-3位羰基的α-位上形成-co-r2取代基,获得中间体(4);
优选地,在步骤c)中,所述反应是将中间体(3)溶于四氢呋喃(thf)后冷却至0℃以下,加入碱性化合物、酯类化合物,在室温下搅拌混合反应,将获得的反应产物加水进行淬灭反应,经萃取、洗涤、干燥、过滤、浓缩、分离纯化后,获得中间体(4),所述中间体(4)中,r2具有如式8化合物中相同的定义。
更优选地,所述中间体(3)加入的质量(mg)与四氢呋喃加入的体积(ml)之比为250-350:15-25。进一步优选地,所述中间体(3)加入的质量(mg)与四氢呋喃加入的体积(ml)之比为300:20。
更优选地,所述碱性化合物为甲醇钠。
更优选地,所述中间体(3)与碱性化合物加入的摩尔比为1:1-2。进一步优选地,所述中间体(3)与碱性化合物加入的摩尔比为1:1.2。
更优选地,所述酯类化合物选自甲酸乙酯、乙酸乙酯、三氟乙酸乙酯中的一种。
更优选地,所述中间体(3)与酯类化合物加入的摩尔比为1:1-2。进一步优选地,所述中间体(3)与酯类化合物加入的摩尔比为1:1.0-1.5。
更优选地,所述搅拌时间为3-5h。进一步优选地,所述搅拌时间为4h。
更优选地,所述淬灭反应是通过加水去除反应产物中不需要参与后续反应的溶于水的物质。
更优选地,所述萃取的反应条件为:萃取试剂为乙酸乙酯,萃取次数为3-4次,萃取试剂用量为25-35ml。进一步优选地,所述萃取的反应条件为:萃取试剂为乙酸乙酯,萃取次数为3次,萃取试剂用量为30ml。
更优选地,所述洗涤依次采用水、食盐进行多次洗涤。
更优选地,所述干燥采用无水硫酸钠进行干燥。
优选地,在步骤c)中,所述中间体(4)的产率为65-75%。
d)将中间体(4)与肼类化合物进行缩合反应,使中间体(4)的c-3位羰基脱水形成r2取代基的吡唑杂环,获得中间体(5);
优选地,在步骤d)中,所述缩合反应是将中间体(4)与肼类化合物溶于有机溶剂,加热搅拌反应后冷却至室温,将获得的反应产物浓缩、洗涤、分离纯化后,获得中间体(5),所述中间体(5)中,r1和r2具有如式8化合物中相同的定义。
更优选地,所述肼类化合物选自烷基肼、芳基肼、杂芳基肼中的一种。进一步优选地,所述肼类化合物选自苯肼、对甲苯肼、对氯苯肼、对溴苯肼、对羧基苯肼、对氰基苯肼中的一种。
更选地,所述中间体(4)与肼类化合物加入的摩尔比为1:1-2。进一步优选地,所述中间体(4)与肼类化合物加入的摩尔比为1:1。
更优选地,所述有机溶剂为乙醇。
更优选地,所述中间体(4)加入的质量(mg)与有机溶剂加入的体积(ml)之比为310-330:15-25。进一步优选地,所述中间体(4)加入的质量(mg)与有机溶剂加入的体积(ml)之比为320:20。
更优选地,所述加热搅拌反应的条件为:加热温度为80-90℃,搅拌时间为11-13h。
进一步优选地,所述加热搅拌反应的条件为:加热温度为85℃,搅拌时间为12h。
更优选地,所述洗涤采用水进行多次洗涤。
优选地,在步骤d)中,所述中间体(5)的产率为80-90%。
e)将中间体(5)进行氢化反应,使中间体(5)脱出c-28位羧基上的苄基保护基,获得中间体(6);
优选地,在步骤e)中,所述氢化反应是将中间体(5)和催化剂溶于有机溶剂,再通入氢气进行反应,将获得的反应产物过滤、洗涤、浓缩后搅拌打浆,再次过滤、干燥,即得所需中间体(6),所述中间体(6)中,r1和r2具有如式8化合物中相同的定义。
更优选地,所述催化剂为10wt%钯碳混合物(pd与c的重量之比为10:90)。
更优选地,所述中间体(5)与催化剂加入的质量之比为30-40:45-55。进一步优选地,所述中间体(5)与催化剂加入的质量之比为35:50。
更优选地,所述有机溶剂为甲醇。
更优选地,所述中间体(5)加入的质量(mg)与有机溶剂加入的体积(ml)之比为30-40:15-25。进一步优选地,所述中间体(5)加入的质量(mg)与有机溶剂加入的体积(ml)之比为35:20。
更优选地,所述氢气在常压下通入。
更优选地,所述催化氢化反应在常压下进行。
更优选地,所述洗涤采用甲醇进行多次洗涤。
更优选地,所述再次过滤为减压抽滤方式。
优选地,在步骤e)中,所述中间体(6)的产率为85-90%。
f)使中间体(6)中的c-28位羧基酰氯化,获得中间体(7);
优选地,在步骤f)中,所述酰氯化是将中间体(6)溶于无水二氯甲烷中冷却至0℃以下,再依次加入草酰氯、n,n-二甲基酰胺(dmf),在室温下搅拌混合反应,浓缩后干燥,获得中间体(7),所述中间体(7)中,r1和r2具有如式8化合物中相同的定义。
更优选地,所述中间体(6)加入的质量(g)与无水二氯甲烷加入的体积(ml)之比为1:2-10。
更优选地,所述冷却是在冰水浴中进行冷却至0℃以下。
更优选地,所述中间体(6)与草酰氯加入的摩尔比为1:1-2。
更优选地,所述中间体(6)与n,n-二甲基酰胺加入的摩尔比为1:0.01-0.1。
更优选地,所述搅拌时间为5-7h。进一步优选地,所述搅拌时间为6h。
g)使中间体(7)的c-28位羧基酰氯与胺类化合物形成酰胺键,即得所需化合物(8)。
优选地,在步骤g)中,所述形成酰胺键是将中间体(7)溶于n,n-二甲基酰胺(dmf),依次加入碱性化合物、胺类化合物,在室温下搅拌混合反应,浓缩、分离纯化、搅拌打浆后干燥,即得所需化合物(8)。
更优选地,所述中间体(7)加入的质量(g)与n,n-二甲基酰胺(dmf)加入的体积(ml)之比为1:2-10。
更优选地,所述碱性化合物为三乙胺(et3n)或n,n-二异丙基乙胺(ipr2net)。
更优选地,所述中间体(7)与碱性化合物加入的摩尔比为1:1-2。
更优选地,所述胺类化合物的结构通式为:r3r4nh,式中,r3和r4具有如式8化合物中相同的定义。
更优选地,所述中间体(7)与胺类化合物加入的量的摩尔比为1:1-1.5。
更优选地,在步骤e)或g)中,所述搅拌打浆是将残留物经乙醚搅拌打浆。
优选地,在步骤a)、b)、c)或e)中,所述过滤是将反应产物经硅藻土过滤除去不溶物。
优选地,在步骤b)、c)、d)、e)、f)或g)中,所述浓缩是将反应产物进行加压蒸馏浓缩。
优选地,在步骤b)、c)、d)或g)中,所述分离纯化是将反应产物经硅胶柱进行层析分离纯化。
优选地,在步骤a)、e)、f)或g)中,所述干燥为真空干燥,所述真空干燥的条件分别为55-65℃,真空度0.005-0.015pa。所述真空干燥为常规采用油泵进行抽真空的干燥方式。
本发明第三方面提供一类含吡唑杂环的熊果酰胺衍生物在制备用于治疗肿瘤的药物中的用途。
优选地,所述肿瘤为宫颈癌或肺癌。
更优选地,所述肿瘤的细胞为宫颈癌细胞hela或肺癌细胞spc-a-1中的一种或多种。
所述一类含吡唑杂环的熊果酰胺衍生物制备用于治疗肿瘤的药物的机理为对肿瘤细胞进行杀伤,从而抑制其活性,进而抑制肿瘤细胞生长。
本发明第四方面提供一种药物组合物,包括治疗有效量的一类含吡唑杂环的熊果酰胺衍生物。
如上所述,本发明提供的一类含吡唑杂环的熊果酰胺衍生物及其合成与应用,通过在熊果酸a环的结构修饰,合成了一系列在熊果酸a环上具有吡唑杂环的熊果酰胺衍生物。经过体外抗肿瘤的活性研究表明,这类熊果酸三萜类衍生物对肿瘤细胞生长具有明显的抑制作用,具有新型抗肿瘤药物的开发潜力。
附图说明
图1显示为本发明的一类含吡唑杂环的熊果酰胺衍生物在不同时间段对肿瘤细胞hela的抑制活性示意图。
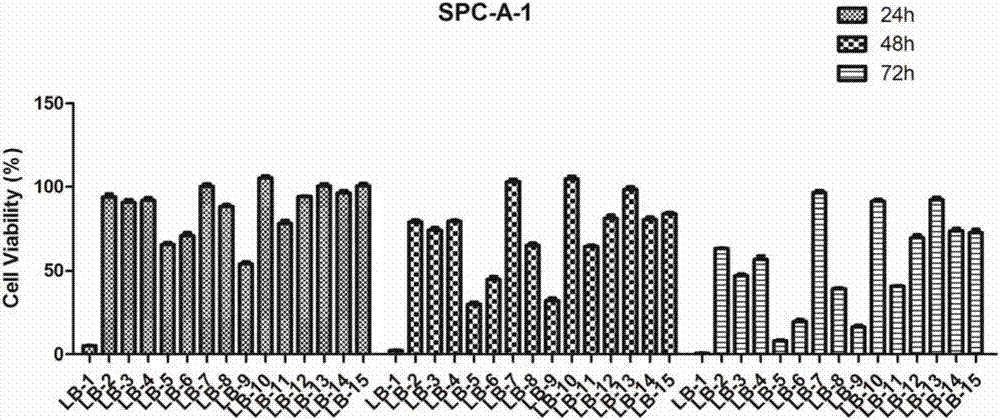
图2显示为本发明的一类含吡唑杂环的熊果酰胺衍生物在不同时间段对肿瘤细胞spc-a-1的抑制活性示意图。
具体实施方式
下面结合具体实施例进一步阐述本发明,应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
以下实施例中使用的试剂均为本领域常规使用的试剂,使用的仪器均为本领域常规使用的仪器。
实施例1
取熊果酸(1)460mg、无水碳酸钾280mg溶于10mln,n-二甲基酰胺中,再加入bnbr300mg,反应混合物在60℃左右加热4小时至反应进行完全。混合物冷却至室温下,加入50mlh2o,析出白色固体。所得固体依次经过滤、水洗、真空干燥即得0.50g中间体(2),中间体(2)的产率为92%。
中间体(2)的谱图鉴定结果如下:
1hnmr(500mhz,cdcl3):δ=7.36-7.29(m,5h),5.24-5.23(m,1h),5.10(d,j=12.5hz,1h),4.98(d,j=12.5hz,1h),3.20(dd,j=11.0,4.5hz,1h),2.04-1.98(m,1h),1.89-1.76(m,3h),1.73-1.67(m,2h),1.64-1.55(m,4h),1.53-1.44(m,6h),1.37-1.26(m,6h),1.07(s,3h),1.05-1.02(m,2h),0.98(s,3h),0.93(d,j=6.0hz,3h),0.89(s,3h),0.85(d,j=6.5hz,3h),0.78(s,3h),0.64(s,3h).
13cnmr(125mhz,cdcl3):δ=177.3,138.1,136.3,128.4(2c),128.1(2c),127.9,125.7,79.0,66.0,55.2,52.9,48.1,47.5,42.0,39.5,39.1,38.8,38.7,38.6,36.9,36.6,33.0,30.6,28.1,27.9,27.2,24.2,23.5,23.2,21.2,18.3,17.0(2c),15.6,15.4.
合成过程如下:
实施例2
取中间体(2)450mg溶于50ml无水二氯甲烷中,冷却至0℃以下,加入pcc355mg反应,混合物缓慢升至室温下搅拌12小时左右至反应进行完全。反应混合物经硅藻土过滤除去不溶物,有机相加压浓缩,残留物经硅胶柱层析分离纯化即得375mg中间体(3),中间体(3)为白色固体,中间体(3)的产率为84%。
中间体(3)的谱图鉴定结果如下:
1hnmr(500mhz,cdcl3):δ=7.37-7.29(m,5h),5.25(t,j=3.5hz,1h),5.11(d,j=12.5hz,1h),4.99(d,j=12.5hz,1h),2.57-2.50(m,1h),2.40-2.34(m,1h),2.27(d,j=11.5hz,1h),2.04-1.98(m,1h),1.92-1.87(m,3h),1.82-1.68(m,3h),1.64-1.54(m,3h),1.50-1.41(m,5h),1.35-1.28(m,4h),1.10-1.07(m,1h),1.08(s,6h),1.03(d,j=9.0hz,6h),0.93(d,j=6.0hz,3h),0.85(d,j=6.5hz,3h),0.68(s,3h).
13cnmr(125mhz,cdcl3):δ=177.2,138.2,136.3,128.4(2c),128.1(2c),127.9,125.4,66.0,55.2,52.9,48.1,47.4,46.7,42.1,39.4,39.3,39.1,38.8,36.6,36.5,34.2,32.5,30.6,27.9,26.5,24.2,23.5,23.3,21.4,21.1,19.5,17.0,16.9,15.2.
合成过程如下:
实施例3
取中间体(3)300mg溶于20ml无水四氢呋喃中,反应液冷却至0℃下,向其中加入甲醇钠45mg,然后加入甲酸乙酯45mg。反应混合物置于室温下搅拌约4小时至反应进行完全。加入少量水淬灭反应,混合物经乙酸乙酯萃取三次(3x30ml)。合并有机相依次用水、食盐水洗、无水硫酸钠干燥、过滤。有机相浓缩,残留经硅胶柱层析快速分离得中间体(4a)。
中间体(4a)的谱图鉴定结果如下:
1hnmr(500mhz,cdcl3):δ=14.92(brs,1h),8.57(s,1h),7.37-7.30(m,5h),5.28(t,j=3.3hz,1h),5.12(d,j=12.5hz,1h),4.99(d,j=12.5,1h),2.32-2.28(m,2h),2.05-1.92(m,4h),1.82-1.69(m,3h),1.65-1.56(m,2h),1.50-1.43(m,3h),1.39-1.26(m,5h),1.18(s,3h),1.14-1.10(m,2h),1.11(s,3h),1.08(s,3h),0.94(d,j=6.5hz,3h),0.89(s,3h),0.86(d,j=6.5hz,3h),0.68(s,3h).
13cnmr(125mhz,cdcl3):δ=177.2,138.1,136.3,128.4(2c),128.2(2c),127.9,125.4,105.8,66.0,53.0,52.0,48.1,45.5,42.2,40.1,39.4,39.3,39.1,38.8,36.6,36.2,32.3,30.6,28.4,27.9,24.2,23.4,23.3,21.1,20.9,19.4,17.0,16.9,14.6.
hrms(esi):m/z[m+h]+calcdforc38h52o4:572.3866;found:572.3871.
合成过程如下:
实施例4
取中间体(3)300mg溶于20ml无水四氢呋喃中,反应液冷却至0℃下,向其中加入甲醇钠45mg,然后加入乙酸乙酯55mg。将反应置于室温下搅拌4小时左右至反应进行完全,加入少量水淬灭反应。混合物用乙酸乙酯萃取三次(3x30ml)。合并有机相依次经水洗、食盐水洗、无水硫酸钠干燥、过滤、滤液经加压蒸馏浓缩,残留物经硅胶柱层析快速分离即得中间体(4b)。
合成过程如下:
实施例5
取中间体(3)300mg溶于20ml无水四氢呋喃中,反应液冷却至0℃下,向其中加入甲醇钠45mg,然后加入三氟乙酸乙酯55mg。将反应置于室温下搅拌4小时左右至反应进行完全,加入少量水淬灭反应。混合物用乙酸乙酯萃取三次(3x30ml)。合并有机相依次经水洗、食盐水洗、无水硫酸钠干燥、过滤、滤液经加压蒸馏浓缩,残留物经硅胶柱层析快速分离即得中间体(4c)。
合成过程如下:
实施例6
取中间体(4a)320mg、4-氰基苯肼盐酸盐95mg溶于20ml乙醇中,加热至80℃下搅拌反应12小时左右至反应进行完全。反应液冷却至室温下,减压蒸馏除去溶剂,残留物加水洗涤数次,初产物进一步经硅胶柱层析纯化得到315mg中间体(5),中间体(5)为白色固体,中间体(5)的产率为84%。
中间体(5)的谱图鉴定结果如下:
1hnmr(500mhz,cdcl3):δ=7.78(d,j=8.5hz,2h),7.55(d,j=8.5hz,2h),7.34(s,1h),7.37-7.31(m,5h),5.32(t,j=3.0hz,1h),5.12(d,j=12.5,1h),4.98(d.j=12.5hz,1h),4.14-4.09(m,1h),2.77(d,j=15.5hz,1h),2.31(d,j=11.0hz,1h),2.15(d,j=15.5hz,1h),2.05-1.99(m,3h),1.90-1.84(m,1h),1.82-1.60(m,4h),1.50-1.47(m,3h),1.39-1.24(m,6h),1.12-1.08(m,1h),1.09(s,3h),1.03(s,3h),1.02(s,3h),0.95(s,3h),0.93(s,3h),0.88(d,j=3.9hz,3h),0.69(s,3h).
13cnmr(125mhz,cdcl3):δ=177.3,146.7,146.5,139.5,138.1,136.4,132.6,130.0,128.5(2c),128.2(2c),128.0,125.7,118.1,115.1,112.9,66.1,58.5,54.6,53.1,48.3,46.4,42.2,39.5,39.2,38.9,38.0,37.1,36.7,34.7,32.6,30.7,29.7,28.0,24.3,23.4,23.3,22.8,21.2,19.2,18.5,17.0,16.9,15.5.
hrms(esi):m/z[m+h]+calcdforc45h55n3o2:669.4294;found:669.4283.
合成过程如下:
实施例7
取中间体(5)35mg、10%pd/c50mg溶于20ml甲醇中,常压催化氢化。待反应进行完全,过滤除去钯碳,甲醇洗涤数次。滤液加压浓缩,所得残留物经乙醚搅拌打浆,过滤,固体真空干燥即得25mg化合物(6),化合物(6)为白色固体,化合物(6)的产率为86%。
化合物(6)的谱图鉴定结果如下:
1hnmr(500mhz,cdcl3):δ=10.95(brs,1h),7.75(d,j=8.5hz,2h),7.52(d,j=8.5hz,2h),7.37(s,1h),5.32(t,j=3.5hz,1h),2.64(d,j=15.0hz,1h),2.23(d,j=11.5hz,1h),2.14(d,j=15.5hz,1h),2.05-1.99(m,3h),1.90-1.84(m,1h),1.72-1.66(m,4h),1.53-1.49(m,3h),1.42-1.26(m,6h),1.15-1.12(m,1h),1.10(s,3h),1.03(s,3h),0.99(s,3h),0.95(d,j=6.5hz,3h),0.93(s,3h),0.89(d,j=6.0hz,3h),0.84(s,3h).
13cnmr(125mhz,cdcl3):δ=183.6,146.6,146.2,139.3,137.8,132.5(2c),129.9(2c),125.6,117.9,115.0,112.9,54.4,52.6,47.9,46.3,42.1,39.4,39.1,38.7,37.9,36.9,36.6,34.6,32.3,30.6,29.5,27.9,24.0,23.4,23.2,22.7,21.1,19.1,16.9,16.7,15.4.
hrms(esi):m/z[m+h]+calcdforc44h63n4o2:579.3825;found:579.3816.
合成过程如下:
实施例8
取中间体(6)100mg溶于20ml无水ch2cl2中,冷却至0℃以下,依次加入(cocl)225μl和催化量的dmf30mg。反应混合物在室温下搅拌6小时,减压蒸除溶剂,残留物经真空干燥即得中间体(7),中间体(7)无需纯化直接进行下一步反应。
合成过程如下:
实施例9化合物lb-1的制备
取中间体(7)100mg溶于10ml无水dmf中,依次加入35μlet3n、0.22mmolr1r2nh2。混合物在室温下搅拌至反应完全。高真空减压浓缩蒸除溶剂,残留物经硅胶柱层析快速纯化,所得产物经甲基叔丁基醚打浆,收集固体经干燥后得102mg化合物lb-1,化合物lb-1为白色固体,化合物lb-1的产率为89%。
化合物lb-1的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.85–7.70(m,2h),7.59–7.49(m,2h),7.38(s,1h),6.79(t,j=5.2hz,1h),5.40(t,j=3.6hz,1h),3.50(dt,j=16.5,5.6hz,1h),3.40–3.23(m,1h),2.73(m,7h),2.17(d,j=15.0hz,1h),2.09(m,2h),2.05–1.95(m,2h),1.95–1.82(m,5h),1.82–1.59(m,3h),1.56–1.37(m,5h),1.36–1.23(m,4h),1.13(s,4h),1.04(d,j=15.8hz,6h),0.99–0.93(m,5h),0.92(d,j=6.5hz,3h),0.90–0.83(m,5h)ppm.
hrms(esi):m/z[m+h]+calcdforc44h62n5o:676.4954;found:676.4957.
合成过程如下:
实施例10化合物lb-2的制备
制备方法同化合物lb-1的制备方法。化合物lb-2的产率为72%。
化合物lb-2的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.93–7.63(m,2h),7.63–7.45(m,2h),7.39(s,1h),5.83(d,j=6.5hz,1h),5.39(t,j=3.6hz,1h),4.12(m,1h),2.67(d,j=14.9hz,1h),2.18(d,j=14.9hz,1h),2.13–2.04(m,2h),2.03–1.82(m,5h),1.81–1.57(m,8h),1.57–1.40(m,7h),1.38–1.28(m,2h),1.14(s,4h),1.04(d,j=12.9hz,6h),0.97(s,7h),0.94–0.82(m,7h)ppm.
hrms(esi):m/z[m+h]+calcdforc43h59n4o:647.4689;found:647.4694.
合成过程如下:
实施例11化合物lb-3的制备
制备方法同化合物lb-1的制备方法。化合物lb-3的产率为72%。
化合物lb-3的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=8.57(d,j=5.0hz,2h),7.78(d,j=8.2hz,2h),7.54(d,j=8.3hz,2h),7.39(s,1h),7.20(d,j=5.0hz,2h),6.34(dt,j=11.5,5.8hz,1h),5.40(t,j=3.5hz,1h),4.62(dt,j=15.5,7.7hz,1h),4.19(dd,j=15.6,5.2hz,1h),2.64(dd,j=15.0,5.6hz,1h),2.21–2.12(m,1h),2.12–1.90(m,6h),1.82(m,1h),1.71(m,2h),1.62–1.47(m,5h),1.41-1.28(m,4h),1.15(s,4h),1.04(d,j=13.7hz,6h),0.99(d,j=5.2hz,3h),0.96–0.89(m,6h),0.76(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc44h56n5o:670.4485;found:670.4494.
合成过程如下:
实施例12化合物lb-4的制备
制备方法同化合物lb-1的制备方法。化合物lb-4的产率为75%。
化合物lb-4的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.81–7.73(m,2h),7.57–7.51(m,2h),7.41–7.35(m,2h),6.33(m,1h),6.27(q,j=5.3hz,1h),6.20(d,j=3.3hz,1h),5.38(t,j=3.5hz,1h),4.44–4.32(m,2h),2.64(dd,j=14.9,6.1hz,1h),2.15(d,j=14.9,5.1hz,1h),2.09–1.97(m,3h),1.97–1.87(m,2h),1.78(m,1h),1.75–1.64(m,3h),1.57–1.46(m,5h),1.44–1.34(m,2h),1.33–1.21(m,2h),1.12(m,4h),1.03(d,j=12.7hz,6h),0.97(d,j=2.6hz,3h),0.93(s,3h),0.90(d,j=6.5hz,3h),0.74(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc43h55n4o2:659.4325;found:659.4327.
合成过程如下:
实施例13化合物lb-5的制备
制备方法同化合物lb-1的制备方法。化合物lb-5的产率为83%。
化合物lb-5的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.79–7.73(m,2h),7.57–7.51(m,2h),7.38(s,1h),5.33(d,j=3.6hz,1h),3.63(d,j=17.4hz,4h),2.65(d,j=15.0hz,1h),2.53–2.49(m,1h),2.43–2.36(m,5h),2.31(s,4h),2.15(d,j=15.2hz,2h),2.11–2.05(m,2h),1.82–1.75(m,2h),1.72(m,2h),1.54–1.49(m,3h),1.42(qd,j=9.7,8.2,4.8hz,3h),1.32–1.25(m,1h),1.12(s,4h),1.09–1.00(m,7h),1.00–0.94(m,6h),0.92(d,j=6.4hz,3h),0.85(d,j=9.3hz,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc43h60n5o:662.4798;found:662.4810.
合成过程如下:
实施例14化合物lb-6的制备
制备方法同化合物lb-1的制备方法。化合物lb-6的产率为72%。
化合物lb-6的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.77(d,j=8.2hz,2h),7.54(d,j=8.2hz,2h),7.38(s,1h),6.43(t,j=5.3hz,1h),5.39(d,j=3.5hz,1h),3.76(t,j=4.7hz,4h),3.45(m,1h),3.11(m,1h),2.70–2.62(m,1h),2.49–2.39(m,6h),2.17(d,j=15.2hz,1h),2.13–1.96(m,4h),1.83(m,1h),1.78–1.66(m,5h),1.51(m,4h),1.42(m,3h),1.31–1.25(m,3h),1.14(s,4h),1.04(d,j=15.4hz,6h),0.97(d,j=12.1hz,6h),0.93(d,j=6.4hz,3h),0.86(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc45h64n5o2:706.5060;found:706.5086.
合成过程如下:
实施例15化合物lb-7的制备
制备方法同化合物lb-1的制备方法。化合物lb-7的产率为78%。
化合物lb-7的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.78(d,j=8.0hz,2h),7.55(d,j=8.1hz,2h),7.39(s,1h),7.25(dd,j=8.5,5.4hz,2h),7.03(t,j=8.5hz,2h),6.17(t,j=5.4hz,1h),5.33(t,j=3.5hz,1h),4.50(dd,j=14.6,5.9hz,1h),4.21(dd,j=14.5,4.9hz,1h),2.69–2.60(m,1h),2.16(d,j=15.0hz,1h),2.10–1.89(m,5h),1.80(m,1h),1.71(m,3h),1.58–1.45(m,5h),1.40(m,2h),1.30(m,2h),1.14(s,4h),1.05(d,j=11.9hz,6h),0.98(s,3h),0.94(s,3h),0.91(d,j=6.3hz,3h),0.76(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc45h56fn4o:687.4438;found:687.4445.
合成过程如下:
实施例16化合物lb-8的制备
制备方法同化合物lb-1的制备方法。化合物lb-8的产率为85%。
化合物lb-8的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.82–7.71(m,2h),7.59–7.50(m,2h),7.40(s,1h),7.19–7.08(m,2h),6.93–6.87(m,2h),5.89(dd,j=7.0,3.7hz,1h),5.04(t,j=3.4hz,1h),3.83(s,3h),3.72(m,1h),3.26–3.11(m,1h),2.82(m,1h),2.71–2.65(m,1h),2.61(d,j=15.0hz,1h),2.14(d,j=15.0hz,1h),1.94(m,3h),1.77-1.59(m,6h),1.54–1.34(m,7h),1.33–1.24(m,4h),1.09(s,4h),1.04(d,j=10.4hz,6h),0.97(s,4h),0.93(s,3h),0.71(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc46h59n4o2:713.4795;found:713.4796.
合成过程如下:
实施例17化合物lb-9的制备
制备方法同化合物lb-1的制备方法。化合物lb-9的产率为81%。
化合物lb-9的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.77(d,j=8.2hz,2h),7.54(d,j=8.2hz,2h),7.39(s,1h),5.97(dd,j=6.9,4.3hz,1h),5.42(d,j=3.5hz,1h),3.28(dt,j=13.4,6.8hz,1h),2.79(m,4.2hz,1h),2.66(d,j=15.0hz,1h),2.18(d,j=15.0hz,1h),2.10(m,2h),2.04–1.85(m,3h),1.82–1.62(m,5h),1.59–1.37(m,7h),1.37–1.22(m,2h),1.14(s,4h),1.04(d,j=13.8hz,6h),0.97(d,j=8.1hz,6h),0.95–0.90(m,9h),0.86(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc42h59n4o:635.4689;found:635.4694.
合成过程如下:
实施例18化合物lb-10的制备
制备方法同化合物lb-1的制备方法。化合物lb-10的产率为76%。
化合物lb-10的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.83–7.71(m,2h),7.60–7.48(m,2h),7.39(s,1h),6.18(dd,j=7.4,4.9hz,1h),5.46(t,j=3.6hz,1h),4.22–4.11(m,1h),3.59(m,1h),2.67(d,j=14.9hz,1h),2.18(d,j=15.0hz,1h),2.11(m,2h),2.08–2.03(m,1h),1.99(m,1h),1.86(m,1h),1.80–1.72(m,2h),1.72–1.63(m,2h),1.60–1.46(m,5h),1.46–1.36(m,2h),1.33–1.26(m,2h),1.15(m,4h),1.04(d,j=14.6hz,6h),0.98(d,j=11.3hz,6h),0.93(d,j=6.4hz,3h),0.84(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc40h52f3n4o:661.4093;found:661.4098.
合成过程如下:
实施例19化合物lb-11的制备
制备方法同化合物lb-1的制备方法。化合物lb-11的产率为82%。
化合物lb-11的谱图鉴定结果如下:
hrms(esi):m/z[m+h]+calcdforc42h57n4o2:649.4482;found:649.4470.
合成过程如下:
实施例20化合物lb-12的制备
制备方法同化合物lb-1的制备方法。化合物lb-12的产率为76%。
化合物lb-12的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.77(d,j=8.0hz,2h),7.54(d,j=8.2hz,2h),7.39(s,1h),6.03(t,j=5.1hz,1h),5.44(d,j=3.7hz,1h),3.22(dt,j=13.0,6.3hz,1h),2.90(ddd,j=13.8,7.6,4.2hz,1h),2.67(d,j=14.9hz,1h),2.18(d,j=15.0hz,1h),2.11(dq,j=7.6,3.8,3.0hz,2h),2.03–1.86(m,3h),1.82–1.65(m,4h),1.61–1.36(m,7h),1.34-1.25(m,2h)1.15(s,4h),1.04(d,j=14.2hz,6h),0.98(d,j=8.6hz,6h),0.93(d,j=6.4hz,3h),0.88(s,4h),0.51(dt,j=8.1,1.9hz,2h),0.26–0.12(m,2h)ppm.
hrms(esi):m/z[m+h]+calcdforc42h57n4o:633.4532;found:633.4539.
合成过程如下:
实施例21化合物lb-13的制备
制备方法同化合物lb-1的制备方法。化合物lb-13的产率为76%。
化合物lb-13的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.86–7.70(m,2h),7.62–7.45(m,2h),7.39(s,1h),5.97–5.84(m,1h),5.41(t,j=3.6hz,1h),3.31(dq,j=13.4,6.8hz,1h),3.07(dtd,j=11.6,7.1,4.4hz,1h),2.66(d,j=14.9hz,1h),2.18(d,j=15.0hz,1h),2.14–2.07(m,2h),1.99(td,j=13.6,4.0hz,1h),1.95–1.85(m,2h),1.82–1.65(m,4h),1.59–1.38(m,10h),1.36–1.28(m,8h),1.14(s,4h),1.04(d,j=14.2hz,6h),0.97(d,j=6.0hz,6h),0.94–0.88(m,6h),0.87(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc44h63n4o:663.5002;found:663.5005.
合成过程如下:
实施例22化合物lb-14的制备
制备方法同化合物lb-1的制备方法。化合物lb-14的产率为76%。
化合物lb-14的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.84–7.68(m,2h),7.61–7.46(m,2h),7.38(s,1h),6.39(t,j=5.1hz,1h),5.39(t,j=3.7hz,1h),3.60–3.52(m,2h),3.50–3.44(m,1h),3.41–3.34(m,1h),3.24(dq,j=13.3,5.7hz,1h),2.66(d,j=15.0hz,1h),2.17(d,j=15.0hz,1h),2.09(m,2h),2.03–1.94(m,2h),1.81(m,2h),1.74(m,4h),1.52(m,4h),1.45–1.37(m,3h),1.20(dd,j=6.1,1.5hz,8h),1.13(s,4h),1.04(d,j=14.9hz,6h),1.00–0.94(m,7h),0.91(d,j=6.5hz,3h),0.87(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc44h63n4o2:679.4951;found:679.4964.
合成过程如下:
实施例23化合物lb-15的制备
制备方法同化合物lb-1的制备方法。化合物lb-15的产率为76%。
化合物lb-14的谱图鉴定结果如下:
1hnmr(500mhz,chloroform-d)δ=7.83–7.71(m,2h),7.60–7.51(m,2h),7.39(s,1h),5.34(d,j=3.8hz,1h),3.47(s,3h),2.65(d,j=15.0hz,1h),2.56(d,j=11.2hz,1h),2.21–1.99(m,4h),1.93–1.69(m,7h),1.56–1.48(m,3h),1.47–1.37(m,3h),1.34-1.24(m,4h)1.13(s,4h),1.04(d,j=15.7hz,6h),0.97(d,j=6.3hz,3h),0.96–0.91(m,6h),0.91–0.85(m,2h),0.85(s,3h)ppm.
hrms(esi):m/z[m+h]+calcdforc42h57n4o:633.4532;found:633.4539.
合成过程如下:
实施例24熊果酰胺衍生物体外抗肿瘤活性
对熊果酰胺衍生物进行了抗肿瘤活性研究,选取肿瘤细胞hela和spc-a-1为检测细胞,以cck-8比色法为检测方法,酶标仪以450nm条件下测其吸光度并计算细胞抑制率。
实验药液的配制:将测试样品用少量的dmso溶解配制成储备液,即按试验最高浓度的1000倍配制储备液。储备液保存于-20℃冰箱中备用。
人宫颈癌细胞(hela)和人肺癌细胞(spc-a-1)的培养:为贴壁生长细胞,常规培养于dmem培养液内(含10%胎牛血清、青链霉素),置于37℃、5%co2培养箱中培养,每隔3-5d传代一次。取对数生长期的肿瘤细胞,调细胞悬液浓度为1-1.5x103个/ml。在96孔板培养板中每孔加细胞悬液100μl,置于37℃、5%co2培养箱中培养24h。
培养24h后,分别按设计加入药液。将测试药液分别加入各孔中,每个浓度设6个平行孔。实验分为药物试验组(分别加入不同测试药物)、对照组(只加培养液和细胞,不加测试药物)和空白组(只加培养液,不加细胞和测试药物)。将加药后的96孔板置于37℃、5%co2培养箱中培养。
药物处理24h/48h/72h后,向每孔加入10μl的cck-8溶液,将培养板置于培养箱内孵育1-4小时。用酶标仪测定各孔在450nm处的od值,计算细胞抑制率。
细胞抑制率(%)=(对照组od值-实验组od值)/(对照组od值-空白孔od值)x100%
测得的细胞抑制率见图1-2、附表2-3,表明这类化合物对hela和spc-a-1细胞的抑制作用。
从上述实验可知:本发明的熊果酰胺衍生物对人宫颈癌细胞(hela)和人肺癌细胞(spc-a-1)有明显的杀伤作用,具有显著的抑制肿瘤细胞活性。
表2.熊果酰胺衍生物对hela细胞的杀伤作用
表3.熊果酰胺衍生物对spc-a-1细胞的杀伤作用
所以,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!
- 5-吡唑酰胺类化合物及其制备方法与应用与制造工艺
- 一种取代的吡啶联吡唑乙酰胺类化合物及其制备方法和应用
- 一种含苯并烯氟菌唑和氟嘧菌酯的杀菌组合物的制作方法
- 一种含有苯并烯氟菌唑的杀菌组合物的制作方法
- 含有苯并烯氟菌唑和啶菌噁唑的杀菌组合物的制作方法
- 一种杀菌组合物及其应用
- 一种取代的吡唑乙酰胺类化合物及其制备方法和应用
- 3-(6-溴-4-氧-4H-色酮)-1-苯基-5-对甲氧基苯基-1,6a-二氢吡咯啉[3,4-c]吡唑-4,6 ...的制作方法
- 5-(4-氟苯基)-N–1-(3-羟基金刚烷基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 铜催化制备1-取代的吡唑并[1,5-c]喹唑啉骨架化合物的方法
- 苯并双(噻二唑)衍生物、包含其的油墨、以及使用了其的有机电子装置的制造方法
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 一种苯并噻咯类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并噻吩并咔唑类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 靛红杂合喹唑啉类化合物合成的靛红衍生物及其在制备抗肿瘤药物中的应用的制造方法与工艺
- 一种杂环衍生物及使用该杂环衍生物的有机发光器件的制造方法与工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 一种含氮杂环衍生物及使用该含氮杂环衍生物的有机发光器件的制造方法与工艺