2‑甲氧基‑5‑香叶基对苯醌衍生物和ScabelloneC的制备方法与流程
本发明属于药物合成技术领域,具体涉及一种2-甲氧基-5-香叶基对苯醌衍生物的制备方法;还具体涉及一种scabellonec的制备方法。
背景技术:
aplidium属中的海鞘类生物被发现其能够提供多种具有重要生物活性的天然产物而闻名。其结构特征主要为醌类或对苯二酚,而scabellones就属于其中的一类天然产物;aplidiumscabellone这一物种是michaelsen课题组与1924年发现的,在2011年由brentr.copp课题组从其将scabellones这一类天然产物分离出来,这类天然产物表现出对人体的嗜中性粒细胞的呼吸爆发的良好抑制作用。并且scabelloneb具有良好的抗疟生物活性;
目前对scabellonea-c的合成已有一例报道,该报道见一下文献:
(susannat.s.chan,a.norriepearce,anah.januario,michaelj.page,marcelkaiser,renej.mclaughlin,jacquiel.harper,victorial.webb,davidbarker,andbrentr.copp*anti-inflammatoryandantimalarialmeroterpenoidsfromthenewzealandascidianaplidiumscabellonej.org.chem.2011,76(21),pp9151–9156;susannat.s.chan,michaela.pullar,imanm.khalil,emmanuelleallouche,davidbarker,brentr.copp*bio-inspireddimerisationofprenylatedquinonesdirectedtowardsthesynthesisofthemeroterpenoidnaturalproducts,thescabellonestetrahedronletters56,2015,pages1486–1488)。
该报道的合成策略:一、以2,4-二甲氧基苯甲醛为起始原料,通过baeyer—villiger氧化反应得到2,4-二甲氧基苯酚,随后与香叶基溴在氢化钠的条件下得到2-香叶基-4,6-二甲氧基苯酚,通过硝酸铈铵氧化成醌再由连二亚硫酸钠还原得到2-香叶基-6-甲氧基-1,4-对苯二酚。
二、得到单体2-香叶基-6-甲氧基-1,4-对苯二酚后,将其溶解吡啶中,在氧气的参与下室温反应30min,可以得到scabellonea(11%)、scabelloneb(3%),但没有合成scabellonec。
虽然这篇报道是对scabellonea、scabelloneb的首次全合成,但对于合成步骤有些繁琐且总产率不到2%,并且未能成功的合成化合物scabellonec,所以依然存在不足。
然而发明人便想出,先制备得到与scabellonea、scabelloneb、scabellonec结构相近的单体二醌,再通过单体二醌进一步分别制备scabellonea、scabelloneb、scabellonec。
技术实现要素:
有鉴于此,本发明的主要目的在于供一种2-甲氧基-5-香叶基对苯醌衍生物的制备方法,解决了现有技术中制备过程繁琐复杂的问题;本发明的主要目的还在于提供一种scabellonec的制备方法,解决了现有技术中未能制备出scabellonec的问题。
为达到上述目的,本发明的技术方案是这样实现的:一种2-甲氧基-5-香叶基对苯醌衍生物的制备方法,该方法通过如下步骤实现:
步骤1、将1,2,4-三甲氧基苯溶于四氢呋喃中,搅拌下加入四甲基乙二胺并降温至-10~0℃,再加入正丁基锂,反应1~2h后,再加入三甲基氯硅烷,反应20~40min,之后,进行淬灭、萃取、干燥和柱层析分离,制得1,2,4-三甲氧基-3-三甲基硅基-苯;
步骤2、将步骤1制得的1,2,4-三甲氧基-3-三甲基硅基-苯溶于四氢呋喃中,搅拌下加入四甲基乙二胺并降温至-75~-80℃,再加入正丁基锂,升温至-10~0℃反应1~2h,再加入香叶基溴,反应0.5~1.5h,之后,进行淬灭、萃取、干燥和柱层析分离,制得1,2,4-三甲氧基-6-香叶基-苯;
步骤3、将步骤2制得的1,2,4-三甲氧基-6-香叶基-苯溶于乙腈中并降温至0~-40℃,再将硝酸铈铵溶于2~4ml的水中,制得硝酸铈铵溶液,并将硝酸铈铵溶液加入1,2,4-三甲氧基-6-香叶基-苯溶液中,反应3~8min,之后,进行淬灭、萃取、干燥和柱层析分离,制得2-甲氧基-5-香叶基对苯醌衍生物。
优选地,所述步骤1中,所述1,2,4-三甲氧基苯、四甲基乙二胺、正丁基锂和三甲基氯硅烷之间的摩尔比为1:(1.5~2.5):(1~1.5):(1.5~2)。
优选地,所述步骤2中,所述1,2,4-三甲氧基-3-三甲基硅基-苯、四甲基乙二胺、正丁基锂和香叶基溴的摩尔比为1:(2~2.5):(1~1.5):(1.5~2)。
优选地,所述步骤3中,所述1,2,4-三甲氧基-6-香叶基-苯与硝酸铈铵的摩尔为1:(4~5)。
本发明的另一个技术方案是这样实现的:一种scabellonec的制备方法,该方法具体为:
将上述的2-甲氧基-5-香叶基对苯醌衍生物溶于有机溶剂中,在0~25℃下搅拌反应3~4d,旋干,干燥后经柱层析分离,制得scabellonec。
优选地,所述有机溶剂为二氯甲烷、氘代氯仿中的一种或两种。
本发明的另一个技术方案是这样实现的:一种scabellonec的制备方法,该方法具体为:
将上述的2-甲氧基-5-香叶基对苯醌衍生物溶于有机溶剂中,在0~25℃下加入cucl,搅拌反应2~3h,饱和氯化铵淬灭,萃取、洗涤、干燥,干燥后用柱层析分离,制得scabellonec。
优选地,所述有机溶剂为二氯甲烷和吡啶中的一种或两种。
优选地,所述2-甲氧基-5-香叶基对苯醌衍生物与cucl的摩尔比为1:(0.02~0.05)。
与现有技术相比,本发明通过以1,2,4-三甲氧基苯为原料,顺利实现了在常用有机溶剂条件下,通过硝酸铈铵氧化一步得到2-甲氧基-5-香叶基对苯醌衍生物(偶联产物),再通过选用不同的有机溶剂等条件合成了scabellonec,实现了现有技术在制备scabellonec方面零产率的突破,同时采用本发明的制备方法,制得scabellonec的产率也很高。
附图说明
图1为本发明实施例1所得产物的核磁共振氢谱图;
图2为本发明实施例11所得产物的核磁共振氢谱图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明提供了一种2-甲氧基-5-香叶基对苯醌衍生物的制备方法,其反应结构式如下:
反应结构式中:碱1、2为氢化金属碱,烷基金属碱,烷基氨基金属碱。我们试用了正丁基锂、叔丁基锂、氢化钠最终正丁基锂产率更高因而选用正丁基锂,并且原料加入碱后会形成白色悬浊液,所以加入四甲基乙二胺(tmeda)增强拔氢的能力;反应结构式中,氧化剂为硝酸铈铵(can)。
该方法通过如下步骤实现:
步骤1、将1,2,4-三甲氧基苯(化合物1)溶于四氢呋喃中,搅拌下加入四甲基乙二胺并降温至-10~0℃,再加入正丁基锂,反应1~2h后,再加入三甲基氯硅烷,反应20~40min,之后,进行淬灭、萃取、干燥和柱层析分离,制得1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2);
其中,1,2,4-三甲氧基苯(化合物1)、四甲基乙二胺、正丁基锂和三甲基氯硅烷之间的摩尔比为1:(1.5~2.5):(1~1.5):(1.5~2);
步骤2、将步骤1制得的1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)溶于四氢呋喃中,搅拌下加入四甲基乙二胺并降温至-75~-80℃,再加入正丁基锂,升温至-10~0℃反应1~2h,再加入香叶基溴,反应0.5~1.5h,之后,进行淬灭、萃取、干燥和柱层析分离,制得1,2,4-三甲氧基-6-香叶基-苯(化合物3);
其中,1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)、四甲基乙二胺、正丁基锂和香叶基溴的摩尔比为1:(2~2.5):(1~1.5):(1.5~2);
步骤3、将步骤2制得的1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶于乙腈中并降温至0~-40℃,再将硝酸铈铵溶于2~4ml的水中,制得硝酸铈铵溶液,并将硝酸铈铵溶液加入1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶液中,反应3~8min,之后,进行淬灭、萃取、干燥和柱层析分离,制得2-甲氧基-5-香叶基对苯醌衍生物(化合物4);
其中,1,2,4-三甲氧基-6-香叶基-苯(化合物3)与氧化剂的摩尔比为1:(4~5)。
其中,2-甲氧基-5-香叶基对苯醌衍生物还可命名为:6,6'-bis((z)-3,7-dimethylocta-2,6-dienyl)-4,4'-dimethoxy-1,1'-bi(cyclohexa-3,6-diene)-2,2',5,5'-tetraone,且以下的6,6'-bis(3,7-dimethylocta-2,6-dienyl)-4,4'-dimethoxy-1,1'-bi(cyclohexa-3,6-diene)-2,2',5,5'-tetraone均用化合物4代替。
本发明还提供了一种scabellonec的制备方法,其反应结构式如下:
其中,scabellonec的制备方法具体为:
将上述的化合物4溶于二氯甲烷或氘代氯仿中,在0~25℃下搅拌反应3~4d,旋干,干燥后经柱层析分离饱和氯化铵淬灭,萃取、洗涤、干燥,干燥后用柱层析分离,制得scabellonec。
本发明还提供了另一种scabellonec的制备方法,其反应结构式如下:
其中,scabellonec的制备方法具体为:
将上述的化合物4溶于二氯甲烷、吡啶或二氯甲烷和吡啶的混合物中,在0~25℃下加入cucl,搅拌反应2~3h,饱和氯化铵淬灭,萃取、洗涤、干燥,干燥后用柱层析分离,制得scabellonec。
其中,有机溶剂为二氯甲烷和吡啶中的一种或多种;化合物4与cucl的摩尔比为1:(0.02~0.05)。
与现有技术相比,本发明通过以1,2,4-三甲氧基苯为原料,顺利实现了在常用有机溶剂条件下,通过硝酸铈铵氧化一步得到化合物4(2-甲氧基-5-香叶基对苯醌衍生物,学名为:6,6'-bis(3,7-dimethylocta-2,6-dienyl)-4,4'-dimethoxy-1,1'-bi(cyclohexa-3,6-diene)-2,2',5,5'-tetraone),再通过选用不同的有机溶剂等条件合成了scabellonec,实现了现有技术在制备scabellonec方面零产率的突破,同时采用本发明的制备方法,制得scabellonec的产率也很高。
化合物4的制备方法,该方法通过如下步骤实现:
实施例1
步骤1、在氩气保护下,将23.8mmol1,2,4-三甲氧基苯(化合物1)溶于80ml四氢呋喃中,搅拌中滴加47.6mmol四甲基乙二胺(tmeda),降温至0℃,滴加28.5mmol正丁基锂,反应1.5小时后,滴加35.7mmol三甲基氯硅烷(tmscl),反应30min,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:50),制得1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2),产率96%;
步骤2、在氩气的保护下,将上述4.16mmol1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)溶于20ml四氢呋喃中,搅拌下加入8.32mmol四甲基乙二胺并降温至-75~-80℃,再加入5.5mmol正丁基锂,升温至0℃反应1.5h,再加入6.24mmol香叶基溴,搅拌反应1h,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,旋干后用柱层析分离(纯石油醚),在使用柱层析分离式,会有部分产物直接脱掉tms,得到1,2,4-三甲氧基-6-香叶基-苯(化合物3),未反应的产物溶于30ml乙酸乙酯,0℃下加入10.4mmolki和14.56mmol四甲基乙二胺(tmscl)反应4h,加饱和碳酸氢钠淬灭,乙酸乙酯萃取,饱和亚硫酸氢钠洗,加入无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:40)两步产率为72%;
步骤3、将0.65mmol1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶于8ml乙腈中并降温至-30℃,再将2.29mmol硝酸铈铵溶于2ml水中,制得硝酸铈铵溶液,并将硝酸铈铵溶液加入1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶液中,反应5min,乙酸乙酯萃取,饱和食盐水洗3遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:10),制得化合物4,产率为40%。
实施例2
步骤1、在氩气保护下,将23.8mmol1,2,4-三甲氧基苯(化合物1)溶于80ml四氢呋喃中,搅拌中滴加47.6mmol四甲基乙二胺(tmeda),降温至0℃,滴加28.5mmol正丁基锂,反应1.5小时后,滴加35.7mmol三甲基氯硅烷(tmscl),反应30min,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:50),制得1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2),产率96%;
步骤2、在氩气的保护下,将上述4.16mmol1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)溶于20ml四氢呋喃中,搅拌下加入8.32mmol四甲基乙二胺并降温至-75~-80℃,再加入5.5mmol正丁基锂,升温至0℃反应1.5h,再加入6.24mmol香叶基溴,搅拌反应1h,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,旋干后用柱层析分离(纯石油醚),在使用柱层析分离式,会有部分产物直接脱掉tms,得到1,2,4-三甲氧基-6-香叶基-苯(化合物3),未反应的产物溶于30ml乙酸乙酯,0℃下加入10.4mmolki和14.56mmol四甲基乙二胺(tmscl)反应4h,加饱和碳酸氢钠淬灭,乙酸乙酯萃取,饱和亚硫酸氢钠洗,加入无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:40)两步产率为72%;
步骤3、将0.65mmol1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶于8ml乙腈中并降温至0℃,再将2.29mmol硝酸铈铵溶于4ml水中,制得硝酸铈铵溶液,并将氧化剂溶液加入1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶液中,反应5min,乙酸乙酯萃取,饱和食盐水洗3遍,加入无水硫酸钠干燥,经柱层析分离(乙酸乙酯与石油醚的体积比为1:10),制得化合物4,产率为25%。
实施例3
步骤1、在氩气保护下,将23.8mmol1,2,4-三甲氧基苯(化合物1)溶于80ml四氢呋喃中,搅拌中滴加47.6mmol四甲基乙二胺(tmeda),降温至0℃,滴加28.5mmol正丁基锂,反应1.5小时后,滴加35.7mmol三甲基氯硅烷(tmscl),反应30min,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:50),制得1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2),产率96%;
步骤2、在氩气的保护下,将上述4.16mmol1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)溶于20ml四氢呋喃中,搅拌下加入8.32mmol四甲基乙二胺并降温至--75~-80℃,再加入5.5mmol正丁基锂,升温至0℃反应1.5h,再加入6.24mmol香叶基溴,搅拌反应1h,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,旋干后用柱层析分离(纯石油醚),在使用柱层析分离式,会有部分产物直接脱掉tms,得到1,2,4-三甲氧基-6-香叶基-苯(化合物3),未反应的产物溶于30ml乙酸乙酯,0℃下加入10.4mmolki和14.56mmol四甲基乙二胺(tmscl)反应4h,加饱和碳酸氢钠淬灭,乙酸乙酯萃取,饱和亚硫酸氢钠洗,加入无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:40)两步产率为72%;
步骤3、将0.65mmol1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶于8ml乙腈中并降温至0℃,再将2.29mmol硝酸铈铵溶于2ml水中,制得硝酸铈铵溶液,并将氧化剂溶液加入1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶液中,反应5min,乙酸乙酯萃取,饱和食盐水洗3遍,加入无水硫酸钠干燥,经柱层析分离(乙酸乙酯与石油醚的体积比为1:10),制得化合物4,产率为30%。
实施例4
步骤1、在氩气保护下,将23.8mmol1,2,4-三甲氧基苯(化合物1)溶于80ml四氢呋喃中,搅拌中滴加35.7mmol四甲基乙二胺(tmeda),降温至0℃,滴加23.8mmol正丁基锂,反应1.5小时后,滴加42.84mmol三甲基氯硅烷(tmscl),反应30min,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:50),制得1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2),产率80%;
步骤2、在氩气的保护下,将上述4.16mmol1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)溶于20ml四氢呋喃中,搅拌下加入9.15mmol四甲基乙二胺并降温至-75~-80℃,再加入4.16mmol正丁基锂,升温至0℃反应1.5h,再加入7.07mmol香叶基溴,搅拌反应1h,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,旋干后用柱层析分离(纯石油醚),在使用柱层析分离式,会有部分产物直接脱掉tms,得到1,2,4-三甲氧基-6-香叶基-苯(化合物3),未反应的产物溶于30ml乙酸乙酯,0℃下加入10.4mmolki和14.56mmol四甲基乙二胺(tmscl)反应4h,加饱和碳酸氢钠淬灭,乙酸乙酯萃取,饱和亚硫酸氢钠洗,加入无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:40)两步产率为60%;
步骤3、将0.65mmol1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶于8ml乙腈中并降温至0℃,再将2.6mmol硝酸铈铵溶于2ml水中,制得硝酸铈铵溶液,并将氧化剂溶液加入1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶液中,反应5min,乙酸乙酯萃取,饱和食盐水洗3遍,加入无水硫酸钠干燥,经柱层析分离(乙酸乙酯与石油醚的体积比为1:10),制得化合物4,产率为20%。
实施例5
步骤1、在氩气保护下,将23.8mmol1,2,4-三甲氧基苯(化合物1)溶于80ml四氢呋喃中,搅拌中滴加59.5mmol四甲基乙二胺(tmeda),降温至0℃,滴加35.7mmol正丁基锂,反应1.5小时后,滴加47.6mmol三甲基氯硅烷(tmscl),反应30min,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:50),制得1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2),产率85%;
步骤2、在氩气的保护下,将上述4.16mmol1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)溶于20ml四氢呋喃中,搅拌下加入10.4mmol四甲基乙二胺并降温至-75~-80℃,再加入6.24mmol正丁基锂,升温至0℃反应1.5h,再加入8.32mmol香叶基溴,搅拌反应1h,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,旋干后用柱层析分离(纯石油醚),在使用柱层析分离式,会有部分产物直接脱掉tms,得到1,2,4-三甲氧基-6-香叶基-苯(化合物3),未反应的产物溶于30ml乙酸乙酯,0℃下加入10.4mmolki和14.56mmol四甲基乙二胺(tmscl)反应4h,加饱和碳酸氢钠淬灭,乙酸乙酯萃取,饱和亚硫酸氢钠洗,加入无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:40)两步产率为55%;
步骤3、将0.65mmol1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶于8ml乙腈中并降温至0℃,再将3.25mmol硝酸铈铵溶于2ml水中,制得硝酸铈铵溶液,并将氧化剂溶液加入1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶液中,反应5min,乙酸乙酯萃取,饱和食盐水洗3遍,加入无水硫酸钠干燥,经柱层析分离(乙酸乙酯与石油醚的体积比为1:10),制得化合物4,产率为22%。
实施例6
步骤1、在氩气保护下,将23.8mmol1,2,4-三甲氧基苯(化合物1)溶于80ml四氢呋喃中,搅拌中滴加47.6mmol四甲基乙二胺(tmeda),降温至0℃,滴加28.5mmol正丁基锂,反应1小时后,滴加35.7mmol三甲基氯硅烷(tmscl),反应20min,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:50),制得1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2),产率92%;
步骤2、在氩气的保护下,将上述4.16mmol1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)溶于20ml四氢呋喃中,搅拌下加入8.32mmol四甲基乙二胺并降温至-75~-80℃,再加入5.5mmol正丁基锂,升温至-10℃反应1h,再加入6.24mmol香叶基溴,搅拌反应0.5h,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,旋干后用柱层析分离(纯石油醚),在使用柱层析分离式,会有部分产物直接脱掉tms,得到1,2,4-三甲氧基-6-香叶基-苯(化合物3),未反应的产物溶于30ml乙酸乙酯,0℃下加入10.4mmolki和14.56mmol四甲基乙二胺(tmscl)反应4h,加饱和碳酸氢钠淬灭,乙酸乙酯萃取,饱和亚硫酸氢钠洗,加入无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:40)两步产率为62%;
步骤3、将0.65mmol1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶于8ml乙腈中并降温至-30℃,再将2.29mmol硝酸铈铵溶于2ml水中,制得硝酸铈铵溶液,并将硝酸铈铵溶液加入1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶液中,反应3min,乙酸乙酯萃取,饱和食盐水洗3遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:10),制得化合物4,产率为36%。
实施例7
步骤1、在氩气保护下,将23.8mmol1,2,4-三甲氧基苯(化合物1)溶于80ml四氢呋喃中,搅拌中滴加47.6mmol四甲基乙二胺(tmeda),降温至0℃,滴加28.5mmol正丁基锂,反应2小时后,滴加35.7mmol三甲基氯硅烷(tmscl),反应40min,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:50),制得1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2),产率92%;
步骤2、在氩气的保护下,将上述4.16mmol1,2,4-三甲氧基-3-三甲基硅基-苯(化合物2)溶于20ml四氢呋喃中,搅拌下加入8.32mmol四甲基乙二胺并降温至-75~-80℃,再加入5.5mmol正丁基锂,升温至-10℃反应2h,再加入6.24mmol香叶基溴,搅拌反应1.5h,饱和氯化铵淬灭,乙酸乙酯萃取三遍,加入无水硫酸钠干燥,旋干后用柱层析分离(纯石油醚),在使用柱层析分离式,会有部分产物直接脱掉tms,得到1,2,4-三甲氧基-6-香叶基-苯(化合物3),未反应的产物溶于30ml乙酸乙酯,0℃下加入10.4mmolki和14.56mmol四甲基乙二胺(tmscl)反应4h,加饱和碳酸氢钠淬灭,乙酸乙酯萃取,饱和亚硫酸氢钠洗,加入无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:40)两步产率为62%;
步骤3、将0.65mmol1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶于8ml乙腈中并降温至-30℃,再将2.29mmol硝酸铈铵溶于2ml水中,制得硝酸铈铵溶液,并将硝酸铈铵溶液加入1,2,4-三甲氧基-6-香叶基-苯(化合物3)溶液中,反应8min,乙酸乙酯萃取,饱和食盐水洗3遍,加入无水硫酸钠干燥,并用柱层析分离(乙酸乙酯与石油醚的体积比为1:10),制得化合物4,产率为38%。
第一种scabellonec的制备方法,该方法具体为:
实施例8
将0.073mmol化合物4溶于8ml二氯甲烷,室温下搅拌3d,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率50%。
实施例9
将0.073mmol化合物4溶于8ml二氯甲烷,室温下搅拌4d,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率48%。
实施例10
将0.073mmol化合物4溶于8ml二氯甲烷,加入等质量硅胶旋干,室温下搅拌反应3d,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率25%。
实施例11
将0.073mmol化合物4溶于8ml氘代氯仿,室温下搅拌反应3天,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率56%。
实施例12
将0.073mmol化合物4溶于8ml氘代氯仿,室温下搅拌反应4d,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率55%。
第二种scabellonec的制备方法,该方法具体为:
实施例13
将0.073mmol化合物4溶于8ml吡啶,并加入0.146mmol%cucl,室温下搅拌反应2h,加饱和氯化铵淬灭,乙酸乙酯萃取,无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率40%。
实施例14
将0.073mmol化合物4溶于8ml吡啶,并加入0.365mmol%cucl,室温下搅拌反应2h,加饱和氯化铵淬灭,乙酸乙酯萃取,无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率60%。
实施例15
将0.073mmol化合物4溶于8ml二氯甲烷,加入0.365mmol%吡啶和0.365mmol%cucl,室温下搅拌反应2h,加饱和氯化铵淬灭,二氯甲烷萃取,无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率60%。
实施例16
将0.073mmol化合物4溶于8ml甲醇,加入0.365mmol%吡啶和0.365mmol%cucl,室温下搅拌反应2h,加饱和氯化铵淬灭,二氯甲烷萃取,无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率50%。
实施例17
将0.073mmol化合物4溶于8ml吡啶(此处吡啶既当溶剂用又当含氮的有机碱用),0℃下加入0.365mmol%cucl,搅拌反应2h,加饱和氯化铵淬灭,乙酸乙酯萃取,无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率60%。
实施例18
将0.073mmol化合物4溶于8ml吡啶(此处吡啶既当溶剂用又当含氮的有机碱用),0℃下加入0.365mmol%cucl,搅拌反应3h,加饱和氯化铵淬灭,乙酸乙酯萃取,无水硫酸钠干燥,旋干,经柱层析分离(乙酸乙酯与石油醚的体积比为1:15)得到scabellonec产率55%。
谱图检测结果及分析如下:
对实施例1、实施例11所得产物进行核磁共振检测,检测数据结果如下:
实施例1所的产物的具体检测数据为:1hnmr(cdcl3,400mhz):δ=5.92(2h,s),4.99(2h,t,j=4hz),4.84(2h,t,j=4hz),3.82(6h,s),3.12(2h,m),2.90(2h,m),1.95(4h,m),1.87(4h,m),1.63(6h,s),1.55(6h,s),1.52(6h,s)ppm;13cnmr(100mhz,cdcl3):δ=185.49,181.23,158.73,143.53,138.60,138.32,131.72,123.91,118.20,107.23,77.48,77.16,76.84,56.51,39.71,26.95,26.50,25.72,17.74,16.37ppm。
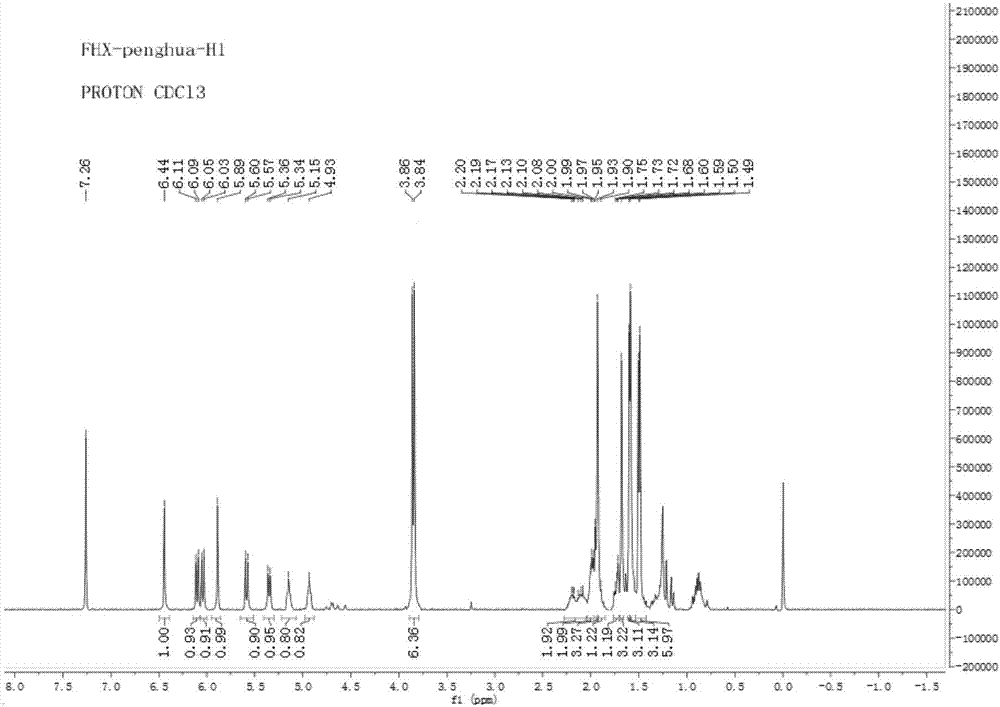
实施例11所的产物的具体检测数据为:1hnmr(cdcl3,400mhz):δ=6.45(1h,s,h-7),6.10(1h,d,j=9.9hz,h-12),6.04(1h,d,j=9.1hz,h-5),5.89(1h,s,h-2),5.58(1h,d,j=9.9hz,h-11),5.35(1h,d,j=9.1hz,h-1′),5.15(1h,t,j=6.7hz,h-3″),4.94(1h,t,j=6.7hz,h-5′),3.86(3h,s,8-och3),3.84(3h,s,3-och3),2.21(1h,m,h-2″a),2.11(1h,m,h-2″b),1.99(2h,m,h2-4′),1.94(2h,m,h2-3′),1.93(3h,s,h3-9′),1.93(1h,m,h-1″a),1.73(1h,m,h-1″b),1.68(3h,s,h3-6″),1.60(3h,s,h3-5″),1.59(3h,s,h3-8′),1.51(3h,s,h3-7′),1.49(3h,s,h3-13)ppm;13cnmr(cdcl3,100mhz):δ=185.5(c-1),179.1(c-4),158.4(c-3),153.1(c-8),151.7(c-6a),144.4(c-2′),138.4(c-8a),134.0(c-12c),131.8(c-6′),131.8(c-4″),131.5(c-4a),126.7(c-11),124.3(c-3″),123.9(c-12),123.7(c-5′),120.3(c-12a),117.1(c-1′),107.7(c-12b),107.2(c-2),101.3(c-7),77.9(c-10),67.8(c-5),56.3(3-och3),56.3(8-och3),41.3(c-1″),39.8(c-3′),26.3(c-4′),25.8(c-6″),25.7(c-8′),24.7(c-13),23.3(c-2″),17.9(c-7′),17.8(c-5″),17.3(c-9′)ppm。
通过检测数据可以看出该方法制备得到化合物4、scabellonec中不同化学环境中的h原子出峰位置稳定,无杂质峰,纯度高,分离性好,产率高。
由于实施例2至实施例2任一所得产物的检测数据与实施例1所得产物的检测数据相同,实施例8至实施例18任一所得产物的检测数据与实施例11所得产物的检测数据相同,因此,不再对除实施例1、实施例11任一所得产物进行数据分析。
以上所述,仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!