苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
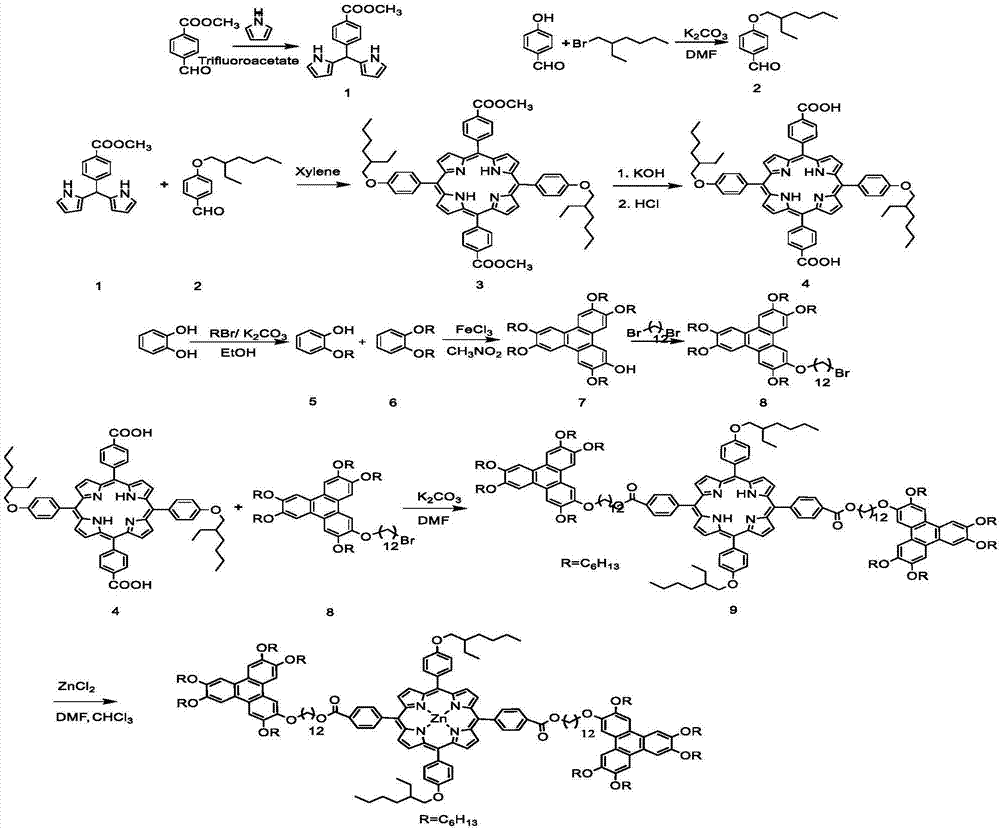
本发明涉及一种苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属zn配合物的合成方法。
背景技术:
苯并菲衍生物是一类非常重要的盘状液晶材料,1978年billard等人首次提出了苯并菲的盘状液晶性质,这一性质引起了科研工作者的极大兴趣,随后一系列苯并菲衍生物应运而生。苯并菲衍生物中柔性侧链的长度影响着物质是否具有液晶性质,过长或过短的柔性侧链都不能使物质显现出液晶性,同时苯并菲衍生物盘状核的有序度随柔性侧链长度的减少而增强,有序度高的盘状液晶化合物易堆集成柱状相,这种柱状相有利于电荷在一维通道上进行传输。在柱状相形成的过程中,电荷的迁移率是逐渐增加的,当盘状液晶化合物完全形成柱状相后,迁移率会突然出现大幅度地增加,这也表明盘状分子排列的有序性增强。
苯并菲衍生物是含有共轭π电子结构的有机化合物,其作为典型的半导体材料受到了广泛的关注,有研究表明,苯并菲类盘状液晶材料已经在静电复印和激光印刷等领域得到了应用,而在有机太阳能电池、有机发光二极管等方面的应用有着很大的发展前景。自1986年邓青云博士制备了第一颗光电转换效率达到1%的双层膜异质结有机太阳能电池后,有机太阳能电池的光电转换效率一直是研究者们关注的热点,电荷传输性能的好坏是制约有机太阳能电池光电转换效率的关键原因之一。而苯并菲类盘状液晶具有的高效电荷传输和能量迁移性能,是制作有机太阳能电池的理想材料之一。
苯并菲类化合物合成的原料易得,产品易于提纯,是一种拥有平面结构的多环芳烃,具有良好的光学性质和热稳定性,因此富含柔性烷氧基侧链的苯并菲类化合物可以作为给体单元;而卟琳类化合物具有高度共轭的大环,在可见光范围内存在吸收光谱,具有良好的光和热稳定性,同时也是很好的电子接受体,且合成方法成熟。在本发明中将苯并菲衍生物作为电子给体单元,卟啉衍生物作为电子受体单元,将电子给体单元和电子受体单元通过柔性桥链连接,形成具有光诱导分子内电子转移的性质的盘状液晶三元化合物。此类化合物未来可以应用在有机太阳能电池、有机光伏材料、液晶材料等分子器件中。
技术实现要素:
本发明的目的是提供一种苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属zn配合物的合成方法。
本发明合成路线分为以下三个部分:第一部分先以对甲酰基苯甲酸甲酯和吡咯为原料合成二吡咯,再以对羟基苯甲醛和1-溴代异辛烷为原料合成对异辛烷氧基苯甲醛,然后二吡咯与对异辛烷氧基苯甲醛反应合成卟啉酯,卟啉酯水解、酸化得到卟啉酸;第二部分在三氯化铁的氧化作用下邻己烷氧基苯酚和邻二己烷氧基苯发生偶联反应生成单羟基五己烷氧基苯并菲,再将单羟基五己烷氧基苯并菲与1,12-二溴十二烷为原料合成具有一个ω-溴支链的烷氧基苯并菲;第三部分将上述两部分得到的中间体合成烷氧基桥连苯并菲-卟啉-苯并菲三元化合物后与金属盐在n,n-二甲基甲酰胺和三氯甲烷的混合溶剂中进行反应,进而得到金属配合物。
苯并菲及其衍生物拥有良好的液晶性质,具有一维方向上的电荷传输和能量迁移性能,且合成苯并菲类化合物的原料易得,产品易于提纯;而卟啉衍生物分子性质和结构稳定,具有良好的荧光性和芳香性,同时也是很好的电子接受体,其合成方法成熟,拥有液晶性质。在本发明中将苯并菲衍生物作为电子给体单元,卟啉衍生物作为电子受体单元,将它们通过柔性桥链连接,形成具有光诱导分子内电子转移的性质的盘状液晶三元化合物。此类化合物未来可以应用在有机太阳能电池、有机光伏材料、液晶材料等分子器件中。
附图说明
图1为本发明苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属zn配合物的结构式。
图2为本发明合成路线的化学反应方程式。
图中:r=c6h13。
具体实施方式
实施例:
实施例中所使用的化学试剂和溶剂均为分析纯。
(1)二吡咯(化合物1)的合成:
取0.22毫升三氟乙酸室温下避光滴加至35毫升含有6克对甲酰基苯甲酸甲酯的吡咯溶液中,氮气氛围下滴加30分钟完毕,接着室温下避光反应4小时后,加入400毫升二氯甲烷,反应混合物用100毫升0.1mol/l的氢氧化钠水溶液洗涤三次,再用100毫升双蒸水洗涤三次,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/二氯甲烷,体积比4:1),得到化合物1,7.20克白色固体,产率70.35%。mp:162.1-162.7℃.ir(kbr)νmax(cm-1):1710,1610,1430,1290,802.1hnmr(500mhz,cdcl3)δ:8.00(d,j=8.2hz,4h),7.36-7.26(m,2h),6.74(s,2h),6.26-6.14(m,2h),5.91(s,2h),5.55(s,1h),3.93(s,3h).
(2)对异辛烷氧基苯甲醛(化合物2)的合成:
取2.44克对羟基苯甲醛、5.20克溴代异辛烷、5.52克无水碳酸钾和20毫升n,n-二甲基甲酰胺,氮气氛围下80℃反应24小时后,加入100毫升水,用60毫升乙酸乙酯萃取三次,分出有机层,再用30毫升饱和食盐水洗涤有机层三次,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/二氯甲烷,体积比10:1),得到化合物2,4.30克淡黄色液体,产率91.88%。bp:>300℃.ir(kbr)νmax(cm-1):1610,1380,1260,841.1hnmr(300mhz,cdcl3)δ:9.87(s,1h),7.68(t,j=8.4hz,2h),6.89(d,j=8.4hz,2h),3.99(t,j=6.9hz,2h),2.13-1.98(m,1h),1.33-1.25(m,8h),0.96(t,j=6.9hz,6h).
(3)5,15-二-(4-甲氧碳基苯基)-10,20-二-(4-异辛氧基)苯基卟啉(化合物3)的合成:
取0.28克化合物1、0.23克化合物2、0.07克间硝基苯甲酸和3毫升二甲苯,140℃反应1小时后,冷却至室温,用200-300目硅胶柱层析提纯(淋洗液:石油醚/二氯甲烷,体积比8:1),得到化合物3,0.13克紫色固体,产率26.37%。mp:>200℃.ir(kbr)νmax(cm-1):1720,1280,801.1hnmr(500mhz,cdcl3)δ:8.95(d,j=4.5hz,4h),8.81(d,j=4.5hz,4h),8.47(d,j=8.0hz,4h),8.34(d,j=8.0hz,4h),8.13(d,j=8.0hz,4h),7.32(d,j=8.0hz,4h),4.24-3.90(m,10h),2.13-1.93(m,2h),1.66-1.46(m,16h),1.17-1.06(m,6h),1.15-0.92(m,6h),-2.73(s,2h).
(4)5,15-二-(4-甲酸基苯基)-10,20-二-(4-异辛氧基)苯基卟啉(化合物4)的合成:
取0.1克化合物3加入15毫升四氢呋喃和15毫升甲醇的混合溶液中,再加入4毫升含有0.4克氢氧化钾的水溶液,80℃冷凝回流反应10小时,冷却至室温后,用2mol/l的盐酸溶液酸化至ph为2~3之间,布氏漏斗抽滤,得化合物4,0.09克墨绿色固体,产率92.63%。mp:>200℃.ir(kbr)νmax(cm-1):2960,1720,1600,1260,802.
(5)邻己烷氧基苯酚(化合物5)的合成:
取30克邻苯二酚、45克1-溴代正己烷、60克无水碳酸钾、2.28克碘化钾和300毫升无水乙醇,85℃冷凝回流反应12小时,冷却至室温后抽滤,滤液减压旋蒸除去溶剂,0.5mmhg的减压蒸馏情况下收集84℃的馏分,得化合物5,15.46克无色油状液体,产率29.52%。bp:106±3℃.ir(kbr)νmax(cm-1):1260,930.1hnmr(300mhz,cdcl3)δ:6.95-6.82(m,4h),5.68(s,1h),4.02(t,j=6.6hz,2h),1.83-1.76(m,2h),1.48-1.31(m,6h),0.91(t,j=6.9hz,3h).
(6)邻二己烷氧基苯(化合物6)的合成:
取10克邻苯二酚、45克1-溴代正己烷、37.26克无水碳酸钾、3.32克碘化钾和125毫升无水乙醇,85℃冷凝回流反应60小时,冷却至室温后抽滤,滤液减压旋蒸除去溶剂,0.5mmhg的减压蒸馏情况下收集142℃的馏分,得化合物6,24.53克无色油状液体,产率96.96%。bp:255±3℃.ir(kbr)νmax(cm-1):1250,939.1hnmr(300mhz,cdcl3)δ:6.89(s,4h),3.99(t,j=6.9hz,4h),1.83-1.76(m,4h),1.49-1.31(m,12h),0.90(t,j=6.9hz,6h).
(7)单羟基-五己烷氧基苯并菲(化合物7)的合成:
取1.16克化合物5和3.32克化合物6溶于60毫升二氯甲烷中,经恒压滴液漏斗滴加至80毫升含有12.96克无水三氯化铁和8毫升硝基甲烷的二氯甲烷溶液中,滴加30分钟完毕,控制反应液温度在0~3℃之间反应4小时后,加入30毫升甲醇和60毫升水终止反应,分出有机层,用30毫升二氯甲烷萃取水层三次,收集所有有机层,再用20毫升饱和食盐水洗涤有机层,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/乙酸乙酯,体积比50:1),得到化合物7,1.80克白色固体,产率40.45%。mp:47.7-50.1℃.ir(kbr)νmax(cm-1):3040,1240,837.1hnmr(400mhz,cdcl3)δ:7.96(s,1h),7.83(s,3h),7.82(s,1h),7.77(s,1h),5.91(s,1h),4.31-4.19(m,10h),1.98-1.90(m,10h),1.59-1.40(m,30h),0.96-0.92(m,15h)
(8)溴代十二氧基-五己氧基苯并菲(化合物8)的合成:
取1.19克化合物6、3.15克1,12-二溴十二烷、0.15克四丁基溴化铵、0.36克氢氧化钾、14毫升水和22毫升二氯甲烷,氮气氛围下室温搅拌反应24小时后,分出有机层,用15毫升二氯甲烷萃取水层三次,收集所有有机层,再用15毫升饱和食盐水洗涤有机层,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/乙酸乙酯,体积比100:1),得到化合物7,1.20克乳白色固体,产率75.60%。bp:>300℃.ir(kbr)νmax(cm-1):2930,1620,1260,836.1hnmr(400mhz,cdcl3)δ:7.84(s,6h),4.23(t,j=6.8hz,12h),3.40(t,j=6.8hz,2h),1.98-1.83(m,12h),1.67-1.50(m,16h),1.42-1.29(m,32h),0.93(t,j=6.8hz,15h).
(9)苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉(化合物9)的合成:
取0.20克化合物4、0.54克化合物8、0.08克碳酸钾和12毫升n,n-二甲基甲酰胺溶液,氮气氛围下80℃回流反应24小时后,加入120毫升水,用60毫升乙酸乙酯萃取三次,分出水层和有机层,用20毫升饱和食盐水洗涤有机层,分出有机层,加入无水硫酸钠干燥除水后,减压旋蒸除去溶剂,得到的粗产物用200-300目硅胶柱层析提纯(淋洗液:石油醚/二氯甲烷,体积比5:1),得到化合物9,0.35克紫色固体,产率60.35%。mp:19.4±3℃.ir(kbr)νmax(cm-1):2930,1720,1260.1hnmr(500mhz,cdcl3)δ:8.93(d,j=4.0hz,4h),8.80(d,j=4.0hz,4h),8.46(d,j=8.0hz,4h),8.32(d,j=8.0hz,4h),8.12(d,j=8.0hz,4h),7.85(s,6h),7.84(s,6h),7.30(d,j=8.0hz,4h),4.53(t,j=6.5hz,4h),4.25-4.22(m,24h),4.15(d,j=6.0hz,4h),1.98-1.92(m,26h),1.71-1.40(m,112h),1.09(t,j=7.5hz,6h),1.00(t,j=7.5hz,6h),0.96-0.93(m,30h),-2.77(s,2h).elementalanalysiscalcdforc182h250n4o18(2782):c,78.58,h,9.06,n,2.01,found:c,78.31,h,9.15,n,2.07.
(10)苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属zn配合物(化合物10)的合成:
取0.13克化合物9、0.07克氯化锌溶于20毫升n,n-二甲基甲酰胺和13毫升三氯甲烷的混合溶液中,65℃搅拌反应2.5小时。反应完向反应液中加入250毫升水,分离出有机层,水层用15毫升三氯甲烷分三次萃取,合并有机层,用10毫升饱和食盐水洗涤有机层。得到的有机液用无水硫酸钠干燥,减压旋蒸后得紫色固体粗产物,将粗产物用硅胶柱进一步提纯(淋洗液:石油醚/乙酸乙酯体积比100:1)。得到化合物10紫色固体0.12g,产率90.17%。mp:>300℃.ir(kbr)νmax(cm-1):3460,2860,2930(c-h),1720,1610(c=c),1510,1430,1390,1260(c-o-c),1170.1hnmr(500mhz,cdcl3)δ:9.01(d,j=4.0hz,4h),8.88(d,j=4.0hz,4h),8.45(d,j=8.0hz,4h),8.32(d,j=8.0hz,4h),8.12(d,j=8.0hz,4h),7.78–7.67(m,12h),7.30(d,j=8.0hz,4h),4.53(t,j=6.5hz,4h),4.23–4.12(m,28h),1.98–1.88(m,26h),1.75–1.41(m,112h),1.10(t,j=7.5hz,6h),1.02(t,j=7.5hz,6h),0.97–0.91(m,30h).
- 还没有人留言评论。精彩留言会获得点赞!
- 苯并菲己烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉金属Zn配合物的合成方法
- 苯并菲己烷氧基桥连十二烷氧基苯基卟啉金属Zn配合物的合成方法
- 一种Meso-四(3,4-二羟基苯基)卟啉锌的制备方法
- 碱性氨基酸修饰氨基四苯基卟啉化合物的新用图
- 使用除草4-氨基-3-氯-6-(4-氯-2-氟-3-甲氧基苯基)吡啶-2-羧酸防治水生杂草的方法
- 苯并菲己烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法
- 一种Meso-四(3,4-二羟基苯基)卟啉钴的制备方法
- 沙格雷酯中间体2-[2-(3-甲氧基苯基)乙基]苯酚的合成方法
- 一种基于一氨基苯基取代卟啉纳米材料的二氧化氮气敏传感器的制造方法
- 取代甲氧基烷氧基苯基烷基衍生物作为防腐剂的用途、保藏方法、化合物和组合物与流程
- 苯并菲己烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料合成方法与流程
- 沙格雷酯中间体2-[2-(3-甲氧基苯基)乙基]苯酚的合成方法
- 一种4,5,6-三氯-n-(2,3-二氟苯基)-2-(3-(三氟甲基)苯氧基)烟酰胺合成方法
- 一种甲基苯基乙烯基乙氧基硅烷的制备方法
- (2e, 6e)-2-(3,5-二甲氧基苯基亚甲基)-6-(4-氯苯基亚甲基)环己酮及其衍生物的新应用
- 一种基于3,4-二甲氧基苯基的取代苯甲酰胺新化合物、制备方法及用图
- 一种n-{[2,5-二乙氧基-4-[3-(3-甲氧基-苯基)-脲基甲基]-苯基}-甲磺酰胺新化合物 ...的制作方法
- 一种n-{[2,5-二乙氧基-4-[3-(4-甲氧基-苯基)-脲基甲基]-苯基}-甲磺酰胺新化合物 ...的制作方法
- 杀真菌的3-{苯基[(杂环基甲氧基)亚氨基]甲基}-杂环衍生物的制作方法
- 一种3-(2-硝基苯甲氧基)-1-(4-氯苯基)-1h-吡唑的绿色合成工艺的制作方法
- 杀真菌的3-{苯基[(杂环基甲氧基)亚氨基]甲基}-杂环衍生物的制作方法
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料合成方法与流程
- 苯并菲己烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- N‑[4‑(烷氧基)‑苯磺酰基]‑5‑芳基‑恶唑‑2‑硫酮类神经氨酸酶抑制剂的制造方法与工艺
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料合成方法与流程
- 苯并菲己烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲己烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料合成方法与流程
- 苯并菲己烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲癸烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉二元化合物盘状液晶材料的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉二元化合物盘状液晶材料合成方法与流程
- 苯并菲己烷氧基桥连异辛烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲十二烷氧基桥连异辛烷氧基苯基卟啉金属Zn配合物的合成方法与流程
- 苯并菲十二烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程
- 苯并菲癸烷氧基桥连十二烷氧基苯基卟啉金属Ni配合物的合成方法与流程