一种头孢克肟的精制方法与流程
本发明属于医药技术领域,涉及一种头孢菌素的制备方法,更具体而言,涉及一种头孢克肟的精制方法。
背景技术:
头孢克肟(cefixime),别名世福素(suprax/cefspan)、达力芬、特普宁、诺百优等,是一种口服第三代头孢菌素类抗生素,由日本藤泽(fujisawa)药品工业株式会社开发研制成功,并于1987年9月率先在日本上市,商品名为cefspan,此后逐渐打入美国、欧洲、中国等20余个国家的药品市场,目前在国际上已得到广泛的认可和临床应用。作为一种重要的口服头孢菌素产品,头孢克肟具备了抗菌谱广、抗菌作用强、高效、耐酶、剂量小等优点。
头孢克肟化学名:[6r-[6α,7β(z)]]-7-[[(2-氨基-4-噻唑基)[(羧甲氧基)亚氨基]乙酰基]氨基]-3-乙烯基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]-2-辛烯-2-羧酸;英文名:(6r,7r)-7-(2-(2-amino-4-thiazolyl)gloxylamido)-8-oxo-3-vinyl-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylicacid;分子式:c16h15n5o7s2;分子量:453.452;casno:79350-37-1;性状:头孢克肟为白色至淡黄色结晶性粉末,无味或具轻微特异臭。在甲醇、二甲亚砜中溶解,略溶于丙酮,难溶于乙醇,在水、乙酸乙酯、乙醚、己烷中几乎不溶。ph值一般为2.6~4.1,熔点为218~225℃。
目前较多的报道集中于头孢克肟的合成工艺上。主要的合成方法包括两种:(1)活性酯法:以(z)-2-(2-氨基噻唑-4-基)-2-甲氧羰基甲氧亚氨基乙酸(以下简称mica)为原料,制备相应的苯并噻唑活性酯,再与7-氨基-3-乙烯基头孢烷酸(以下简称7-avca)进行酰化反应合成头孢克肟甲酯,最后经碱水解脱除保护基得到头孢克肟,具体工艺流程如图1所示。
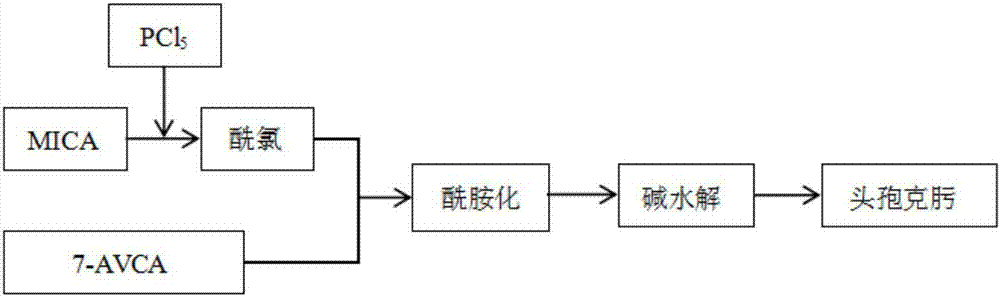
(2)酰氯法:以mica侧链酸为原料,与pcl5反应得到酰氯,再与7-avca酰胺化、经碱水解得到成品,工艺流程如图2所示。
对以上两种合成方法进行对比发现,酰氯法操作较为复杂、且生产过程存在副反应、对环境有较大污染,而活性酯法的原料易得、反应不需要高温条件、所需设备的要求较低,因此常见于工业化大规模生产。
对于头孢克肟的结晶工艺的文献较少,目前中国专利:201010191016.7,201110429323.9与201210217305.9涉及到了头孢克肟的结晶工艺,但是并未涉及在反应结晶中加晶种的方法。
技术实现要素:
为了克服现有技术的缺点与不足,本发明的目的在于提供一种头孢克肟的精制方法。
本发明提供一种生产稳定性好、白度高、粒度分布窄、流动性好、堆密度较大、含量高、产率高、水分与溶残达标的头孢克肟晶体颗粒的新方法。
本发明的目的通过下述技术方案实现:
一种头孢克肟的精制方法,包括如下步骤:
在一定初始浓度的头孢克肟盐有机溶剂与水的混合溶液中或水溶液中,在10~40℃温度下,加入酸,将溶液ph调节到3.25~3.75,同时搅拌,加入一定量晶种,并用在线ph计监控体系ph,5~30min后体系ph开始上升,此时缓慢加酸至终点ph=2.20,降温养晶2~4小时;将产物过滤、洗涤、干燥后得到头孢克肟晶体。
所述的初始浓度为10mg/ml~60mg/ml,优选为20~40mg/ml。
所述的反应体系温度为10~40℃,优选为15~30℃。
所述的酸的种类为:盐酸、硫酸、磷酸、醋酸。
所述的加入酸的质量分数为0.5wt%~8.0wt%,优选为1.0wt%~3.0wt%。
所述的加入酸的速率为0.5ml/min~8ml/min,优选为1.0ml/min~6ml/min。
所述的缓慢加酸的速率为0.5ml/min~8ml/min,优选为1.0ml/min~6ml/min。
所述的加晶种时ph值范围为3.25~3.75,优选为3.35~3.65。
所述的加入晶种量为头孢克肟盐质量的0.5wt%~10wt%,优选为2wt%~8wt%。
所述的养晶时间为:第一次养晶时间为5~30min,优选为10~20min。
所述的搅拌速率为:搅拌速率在加酸阶段为150~350rpm,养晶阶段为80~200rpm,优选的加酸阶段搅拌速率为150~250rpm,优选的养晶阶段搅拌速率为80~150rpm。
头孢克肟稳定性的测试方法采用2015药典规定的高效液相色谱自身对照法测定。
该结晶方法得到的头孢克肟晶体,稳定前最大单杂≤0.15%,稳定前总杂≤0.20%,稳定10天后最大单杂≤0.40%,稳定10天后总杂≤1.50%,白度≥70,粒度:单峰窄分布40~90μm,稳定10天前后含量≥95.0%,水分10%~12%,休止角≤40°。
本发明相对于现有技术具有如下的优点及效果:
(1)利用本发明的方法得到稳定性好、白度高、粒度分布窄、流动性好、堆密度较大、含量高、产率高、水分与溶残达标的头孢克肟晶体颗粒。
(2)实验通过xrd作为产物稳定性预测判据,理论上结晶度越高,稳定性越好。稳定性分为稳定前最大单杂,稳定前总杂,稳定后最大单杂,稳定后总杂。在未自发成核之前加入晶种,可以提高产物的结晶度,从而提高产物的稳定性、白度等各项性能指标。实验的重复性强,受外界因素干扰小,尤其适用于工业生产。
附图说明
图1是活性酯法的具体工艺流程。
图2是酰氯法的工艺流程。
图3是头孢克肟光学显微镜图。
图4是头孢克肟扫描电镜图。
图5是头孢克肟红外光谱图。
图6是头孢克肟dsc、tg图。
图7是头孢克肟粒度分布图。
图8是头孢克肟不同晶种加入量对应产物xrd图谱。
图9是不同头孢克肟盐浓度对应产物xrd图谱。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
头孢克肟光学显微镜图,如图3所示。头孢克肟扫描电镜图,如图4所示。头孢克肟红外光谱图,如图5所示。头孢克肟dsc、tg图,如图6所示。
实施例1
在2l反应釜中加入1500ml初始浓度为10mg/ml的头孢克肟盐溶液,溶剂为以水为主要组分的水、丙酮、乙酸乙酯混合溶剂。反应装置中加装ph计,体系初始ph在5.20左右,搅拌速率为150rpm,待体系温度稳定在10℃±2℃后,开始以0.5ml/min加酸速率加入质量分数为0.5wt%的盐酸;待ph值稳定在3.25左右时,加入0.5wt%的晶种,晶种通过之前制备的产品经过预处理后得到。搅拌5min后,开始以0.5ml/min加酸速率继续加盐酸直到ph值达到滴加终点2.20。将体系温度调整至5℃,搅拌速率调整到80rpm,降温养晶2h。
将产物过滤、洗涤、真空干燥,得到三水合头孢克肟晶体15g。将产物放入西林瓶在药品稳定性加速实验箱中,设定温度60℃稳定10天,分别对稳定前后头孢克肟晶体的稳定性与含量、水分进行分析。
对产品的流动性、粒度分布、白度、溶残值、水分值进行测定。对产品的结晶度进行分析,对产品的外观形貌进行分析。
头孢克肟xrd图谱,如图9所示。
实施例2
与实施例1相同,只是头孢克肟盐初始浓度为20mg/ml。头孢克肟xrd图谱,如图9所示。
实施例3
与实施例1相同,只是头孢克肟盐初始浓度为30mg/ml。
实施例4
与实施例1相同,只是头孢克肟盐初始浓度为40mg/ml。
实施例5
与实施例1相同,只是头孢克肟盐初始浓度为50mg/ml。
实施例6
与实施例1相同,只是头孢克肟盐初始浓度为60mg/ml。
实施例7
与实施例1相同,只是反应体系的溶剂为纯水体系。
实施例8
与实施例1相同,只是反应体系的溶剂为水、丙酮体系。
实施例9
在2l反应釜中加入1500ml初始浓度为40mg/ml的头孢克肟盐溶液,溶剂为以水为主要组分的水、丙酮、乙酸乙酯混合溶剂。反应装置中加装ph计,体系初始ph在5.20左右,搅拌速率为150rpm,待体系温度稳定在20℃±2℃后,开始以0.5ml/min加酸速率加入质量分数为0.5wt%的盐酸;待ph值稳定在3.25左右时,加入0.5wt%的晶种,晶种通过之前产品制备得到。搅拌5min后,开始以0.5ml/min加酸速率继续加盐酸直到ph值达到滴加终点2.20。将体系温度调整至5℃,搅拌速率调整到80rpm,降温养晶2h。
实施例10
与实施例9相同,只是反应体系的温度为15℃±2℃。
实施例11
与实施例9相同,只是反应体系的温度为25℃±2℃。
实施例12
与实施例9相同,只是反应体系的温度为30℃±2℃。
实施例13
与实施例9相同,只是反应体系的温度为35℃±2℃。
实施例14
与实施例9相同,只是反应体系的温度为40℃±2℃。
实施例15
与实施例9相同,只是滴加的酸为磷酸。
实施例16
与实施例9相同,只是滴加的酸为硫酸。
实施例17
与实施例9相同,只是滴加的酸为醋酸。
实施例18
在2l反应釜中加入1500ml初始浓度为40mg/ml的头孢克肟盐溶液,溶剂为以水为主要组分的水、丙酮、乙酸乙酯混合溶剂。反应装置中加装ph计,体系初始ph在5.20左右,搅拌速率为150rpm,待体系温度稳定在20℃±2℃后,开始以0.5ml/min加酸速率加入质量分数为2.0wt%的盐酸;待ph值稳定在3.25左右时,加入2.0wt%的晶种,晶种通过之前产品制备得到。搅拌5min后,开始以0.5ml/min加酸速率继续加盐酸直到ph值达到滴加终点2.20。将体系温度调整至5℃,搅拌速率调整到80rpm,降温养晶2h。
实施例19
与实施例18相同,只是滴加的酸的质量分数为0.5wt%。
实施例20
与实施例18相同,只是滴加的酸的质量分数为1.0wt%。
实施例21
与实施例18相同,只是滴加的酸的质量分数为3.0wt%。
实施例22
与实施例18相同,只是滴加的酸的质量分数为4.0wt%。
实施例23
与实施例18相同,只是滴加的酸的质量分数为5.0wt%。
实施例24
与实施例18相同,只是滴加的酸的质量分数为6.0wt%。
实施例25
与实施例18相同,只是滴加的酸的质量分数为7.0wt%。
实施例26
与实施例18相同,只是滴加的酸的质量分数为8.0wt%。
实施例27
与实施例18相同,只是加酸时搅拌速率为200rpm。
实施例28
与实施例18相同,只是加酸时搅拌速率为250rpm。头孢克肟粒度分布图,如图7所示。
实施例29
与实施例18相同,只是加酸时搅拌速率为300rpm。头孢克肟粒度分布图,如图7所示。
实施例30
与实施例18相同,只是加酸时搅拌速率为350rpm。头孢克肟粒度分布图,如图7所示。
实施例31
与实施例18相同,只是养晶时搅拌速率为100rpm。头孢克肟粒度分布图,如图7所示。
实施例32
与实施例18相同,只是养晶时搅拌速率为150rpm。
实施例33
与实施例18相同,只是晶种加入量为0.5wt%。
将产物放入西林瓶在药品稳定性加速实验箱中,设定温度60℃稳定10天,分别对稳定前后头孢克肟晶体的稳定性与含量、水分进行分析。头孢克肟xrd图谱,如图8所示。
对产品的流动性、粒度分布、白度、溶残值、水分值进行测定。对产品的结晶度进行分析,对产品的外观形貌进行分析。具体结果见表1。
表1头孢克肟产物数据表
实施例34
与实施例18相同,只是晶种加入量为1.0wt%。头孢克肟xrd图谱,如图8所示。
实施例35
与实施例18相同,只是晶种加入量为3.0wt%。头孢克肟xrd图谱,如图8所示。
实施例36
与实施例18相同,只是晶种加入量为4.0wt%。
实施例37
与实施例18相同,只是晶种加入量为5.0wt%。
实施例38
与实施例18相同,只是晶种加入量为6.0wt%。
实施例39
与实施例18相同,只是晶种加入量为7.0wt%。
实施例40
与实施例18相同,只是晶种加入量为8.0wt%。
实施例41
在2l反应釜中加入1500ml初始浓度为40mg/ml的头孢克肟盐溶液,溶剂为以水为主要组分的水、丙酮、乙酸乙酯混合溶剂。反应装置中加装ph计,体系初始ph在5.20左右,搅拌速率为150rpm,待体系温度稳定在20℃±2℃后,开始以0.5ml/min加酸速率加入质量分数为2.0wt%的盐酸;待ph值稳定在3.60左右时,加入2.0wt%的晶种,晶种通过之前产品制备得到。搅拌5min后,开始以0.5ml/min加酸速率继续加盐酸直到ph值达到滴加终点2.20。将体系温度调整至5℃,搅拌速率调整到80rpm,降温养晶2h。
实施例42
与实施例41相同,区别在于待ph稳定在3.35时加入晶种。
实施例43
与实施例41相同,区别在于待ph稳定在3.45时加入晶种。
实施例44
与实施例41相同,区别在于待ph稳定在3.55时加入晶种。
实施例45
与实施例41相同,区别在于待ph稳定在3.65时加入晶种。
实施例46
与实施例41相同,区别在于待ph稳定在3.75时加入晶种。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!