一种DNA甲基化动态过程的高通量在线检测方法与流程
本发明涉及一种dna甲基化动态过程的高通量在线检测方法。
背景技术:
dna甲基化是指dna在甲基转移酶(如dnmt3a)作用下,催化底物s-腺苷甲硫氨酸(sam),转移一个甲基至胞嘧啶的c5位置,生成5-甲基胞嘧啶(5-mc)的过程。作为重要的表观遗传学修饰方式之一,dna甲基化与基因遗传和胚胎发育密切相关,也是肿瘤等疾病的潜在标志物。dna的甲基化修饰方式和程度与mtases的作用方式和活性密切相关。研究dna甲基化机制和分析方法,对于深入理解表观遗传机制、细胞生长过程和重大疾病诊断具有重要意义。
dna甲基化的分析方法分为基因组整体甲基化水平分析,特异位点甲基化分析,以及新甲基化位点寻找等,常规方法包括甲基化特异性pcr(ms-pcr)、甲基化敏感的单核苷酸引物扩增(ms-snupe)、重亚硫酸氢钠联合限制性内切酶分析法(cobra)、甲基化敏感性斑点分析(ms-dba)、限制性标记基因组扫描(rlgs)、甲基化免疫沉淀(mip)等。虽然这些方法已被广泛应用于临床和基础研究,但多数方法操作繁琐、耗时长、检测成本高或依赖复杂设备。更为重要的是,这些方法只能对dna甲基化的存在与否进行检测,无法对dna甲基化的生成过程进行有效分析。实现动态甲基化过程的在线检测,对于加深甲基化过程机制的理解,精确研究甲基化转移酶活性、酶抑制剂、酶序列偏好性等具有重要意义。当前,尚无在线检测动态dna甲基化过程的有效方法。
技术实现要素:
为了解决上述存在的问题,本方法基于甲基转移酶在不同温度下的活性差异,结合特定的控温方式,建立一种快速、简便的dna甲基化动态在线检测方法,同时,利用荧光定量pcr仪实现dna甲基化动态过程的高通量检测。结果表明,在不同温度条件下,甲基转移酶可以对目标dna进行“吸附-脱附”的循环作用,消除非特异性干扰,实现dna甲基化过程的无损在线检测,样本测试值与标准测试值的吻合度为92.07%。
本发明的目的在于提供一种dna甲基化动态过程的高通量在线检测方法。
本发明所采取的技术方案是:
一种dna甲基化动态过程的高通量在线检测方法,包括以下步骤:将荧光标记的目标dna双链、甲基转移酶、甲基转移酶底物和缓冲液的混合物用定量pcr仪进行检测,检测的程序为36~38℃、6~8min,48~50℃、2.5~3.5min;至少10个循环;记录检测过程中的荧光值。
进一步的,步骤1)中,所述dna双链的终浓度为300~800nm。
进一步的,步骤1)中,所述甲基转移酶浓度为100~200nm。
进一步的,步骤1)中,所述甲基转移酶底物的终浓度为100~200μm。
进一步的,荧光标记的目标dna双链中荧光标记的位点情况为:其中一条dna单链的荧光标记在甲基化位点向3’端方向的3bp以内的核苷酸上,另一互补dna序列的荧光标记在3’端,两处标记的荧光相同。
进一步的,步骤1)中,所述缓冲液中含有45~55mmnacl,8~12mmtris-hcl,8~12mmmgcl2,0.8~1.2mmdtt。
进一步的,所述甲基转移酶选自dnmt3a、dnmt3b、dnmt3l、dnmt1中至少一种。
一种甲基化转移酶活性的检测方法,包括以下步骤:将荧光标记的含甲基化位点的dna双链、待测甲基转移酶、甲基转移酶底物和缓冲液的混合物用定量pcr仪进行检测,检测程序为36~38℃、6~8min,48~50℃、2.5~3.5min;至少10个循环;记录检测过程中的荧光值;根据荧光值的变化情况判断甲基化转移酶活性。
一种甲基化转移酶序列偏好性的检测方法,将用荧光标记的待测dna双链序列、待测甲基转移酶、甲基转移酶底物和缓冲液的混合物用定量pcr仪进行检测,检测的程序为36~38℃、6~8min,48~50℃、2.5~3.5min;至少10个循环;记录检测过程中的荧光值;根据荧光值的变化情况,判断甲基化转移酶对不同dna双链序列的偏好性。
一种甲基化转移酶抑制剂的筛选方法,将荧光标记的含甲基化位点的dna双链、甲基转移酶、待测甲基转移酶抑制剂、甲基转移酶底物和缓冲液的混合物用定量pcr仪进行检测,检测的程序为36~38℃、6~8min,48~50℃、2.5~3.5min;至少10个循环;记录检测过程中的荧光值;根据荧光值的变化情况,判断待测甲基转移酶抑制剂是否对甲基化转移酶具有抑制作用。
本发明的有益效果是:
本发明方法通过调控dna甲基化反应的体系温度,调节甲基转移酶的作用状态,使其在特定的时间进行“吸附-脱附”过程,既保证了甲基化反应的持续性,也极大的减少了非特异性干扰。该方法具有快速、便捷、高通量和使用成本低等特点,在dna甲基化检测方面具有独特优势:
(1)建立了dna甲基化过程的无损检测方法,该方法不涉及甲基化以外的任何反应,检测结果最接近待测物的真实状态;
(2)建立了免限制性生物类或荧光类识别物的检测方法,对特异位点的甲基化研究具有普适性;
(3)建立了无需二次标记或二次反应的检测方法,具有操作步骤简化,分析时间短,测定成本低的特点。
附图说明
图1为本发明实验原理示意图;
图2为dna甲基化对fret效果的影响;a为正常dna杂交前后的信号变化;b.甲基化dna杂交前后的信号变化;
图3为甲基转移酶dnmt3a在不同温度下作用所产生的fret信号差异分析;
图4为甲基转移酶dnmt3a吸附时间对fret信号的影响,(甲基转移酶脱附时间3分钟);a.吸附时间3分钟;b.吸附时间5分钟;c.吸附时间7分钟;d.吸附时间9分钟;
图5为甲基转移酶脱附时间对fret信号的影响(甲基转移酶吸附时间7分钟);a.脱附时间1分钟;b.脱附时间3分钟;c.脱附时间5分钟;d.脱附时间7分钟;
图6为dna甲基化过程的在线检测结果;a.单一样本;b.多组分样本;
图7为dna甲基化过程的离线检测。
具体实施方式
一种dna甲基化动态过程的高通量在线检测方法,包括以下步骤:
将荧光标记的目标dna双链、甲基转移酶、甲基转移酶底物和缓冲液的混合物用定量pcr仪进行检测,检测的程序为36~38℃、6~8min,48~50℃、2.5~3.5min;至少10个循环;记录检测过程中的荧光值。
优选的,步骤1)中,所述dna双链的终浓度为300~800nm。
优选的,步骤1)中,所述甲基转移酶浓度为100~200nm。
优选的,步骤1)中,所述甲基转移酶底物的终浓度为100~200μm。
优选的,荧光标记的目标dna双链中荧光标记的位点情况为:其中一条dna单链的荧光标记在甲基化位点向3’端方向的3bp以内的核苷酸上,另一互补dna序列的荧光标记在3’端,两处标记的荧光相同。
优选的,步骤1)中,所述缓冲液中含有45~55mmnacl,8~12mmtris-hcl,8~12mmmgcl2,0.8~1.2mmdtt。
优选的,所述甲基转移酶选自dnmt3a、dnmt3b、dnmt3l、dnmt1中至少一种。
优选的,步骤1)中,所述甲基转移酶底物为s-腺苷甲硫氨酸。
一种甲基化转移酶活性的检测方法,包括以下步骤:将荧光标记的含甲基化位点的dna双链、待测甲基转移酶、甲基转移酶底物和缓冲液的混合物用定量pcr仪进行检测,检测程序为36~38℃、6~8min,48~50℃、2.5~3.5min;至少10个循环;记录检测过程中的荧光值;根据荧光值的变化情况判断甲基化转移酶活性。
优选的,所述缓冲液中含有45~55mmnacl,8~12mmtris-hcl,8~12mmmgcl2,0.8~1.2mmdtt。
一种甲基化转移酶序列偏好性的检测方法,将用荧光标记的待测dna双链序列、待测甲基转移酶、甲基转移酶底物和缓冲液的混合物用定量pcr仪进行检测,检测的程序为36~38℃、6~8min,48~50℃、2.5~3.5min;至少10个循环;记录检测过程中的荧光值;根据荧光值的变化情况,判断甲基化转移酶对不同dna双链序列的偏好性。
优选的,所述缓冲液中含有45~55mmnacl,8~12mmtris-hcl,8~12mmmgcl2,0.8~1.2mmdtt。
一种甲基化转移酶抑制剂的筛选方法,将荧光标记的含甲基化位点的dna双链、甲基转移酶、待测甲基转移酶抑制剂、甲基转移酶底物和缓冲液的混合物用定量pcr仪进行检测,检测的程序为36~38℃、6~8min,48~50℃、2.5~3.5min;至少10个循环;记录检测过程中的荧光值;根据荧光值的变化情况,判断待测甲基转移酶抑制剂是否对甲基化转移酶具有抑制作用。
优选的,所述缓冲液中含有45~55mmnacl,8~12mmtris-hcl,8~12mmmgcl2,0.8~1.2mmdtt。
本发明的实验原理
本发明方案的具体实施步骤如图1所示。以一段包含一个甲基化位点的dna为待测dna。在其甲基化位点的3bp内标记5-羧基荧光基团(5-fam)。设计其互补序列,并在互补序列的3’端标记5-fam。这两条标记5-fam的dna链杂交后,发生荧光共振能量转移(fret),获得初始信号(f0)。在甲基转移酶和底物(如sam)的作用下,发生甲基化反应,降低fret效率,使信号降低。由于甲基转移酶在甲基化dna后,可能没有及时脱离dna双链,使c:g碱基对间的π电子云受到了一定的干扰,引起了fret信号的偏差。升高反应体系温度,使甲基转移酶形态发生改变并从dna双链上脱离。此时fret只受甲基化位点的影响,因此,所获得的信号(f1)与初始信号(f0)的差值可以反映出现阶段目标dna真实的甲基化状态。之后恢复反应体系的温度,使甲基转移酶重新作用于目标dna,继续进行甲基化反应。通过在荧光定量pcr仪上设置温度循环,并记录每一次升温后的fret信号强度变化,可实现dna甲基化过程的高通量在线检测。
本发明方法的构建过程和优化过程
(1)本发明方法的构建过程
1.验证fret效果及dna甲基化对fret效果的影响
将标记有5-fam的dna探针链和互补连混合于磷酸盐缓冲液中,在荧光仪中进行荧光强度测试。之后将该混合液置于在37℃下温浴2小时,进行dna杂交反应。杂交完成后,在荧光仪中进行荧光强度测试。通过对比杂交前后荧光信号的强度变化,验证fret效果。之后,将相同浓度的标记有5-fam的甲基化dna探针链和互补连混合于磷酸盐缓冲液中,重复上述反应和测试。对比两次实验的fret效果差异,验证dna甲基化对fret效果的影响。
2.分析甲基转移酶在不同温度下的作用状态
在反应体系中加入甲基转移酶和底物(如sam),在36~38℃下对目标dna进行甲基化反应。反应进行一段时间后,检测fret信号。之后,升高反应体系温度至48~50℃,并再次检测fret信号。不同的温度将使甲基转移酶呈现不同的反应状态,促使甲基转移酶吸附或脱附于目标dna,fret信号大小也因此发生改变。
(2)本发明方法的优化过程
在反应体系中加入甲基转移酶(如dnmt3a)和底物(如sam),固定反应体系在49℃下的脱附反应时间,并改变其在37℃下的吸附反应时间,分别检测分析不同吸附反应时间所造成的fret信号差异,并确定最佳吸附反应时间。之后,固定反应体系在37℃下的最佳吸附反应时间,并改变其在49℃下的脱附反应时间,分别检测分析不同脱附反应时间所造成的fret信号差异,并确定最佳脱附反应时间。
本发明方法的定量检测过程
单一组分样或多组分样本同时本放置于定量pcr仪中,在反应体系中加入甲基转移酶(如dnmt3a)和底物(如sam)。以最佳的吸附、脱附反应时间为实验条件,调控甲基转移酶目标dna的甲基化反应,并检测fret信号,绘制信号变化曲线。
下面结合具体实施例对本发明作进一步的说明。
实施例1dna甲基化对fret效果的影响
以dna甲基化转移酶dnmt3a作用甲基化位点5’-ccgg-3’为例。
(1)配制500μl样本储备液(s1),其含有500nm标记5-fam的dna探针链、500nm标记5-fam的dna互补链和0.1m磷酸盐缓冲液(ph=7.4)。
(2)配制500μl样本储备液(s2),其含有500nm标记5-fam的甲基化dna探针链(荧光标记在3’端)、500nm标记5-fam的甲基化dna互补链(荧光标记在甲基化位点向3’端方向的3bp以内的核苷酸上)和0.1m磷酸盐缓冲液(ph=7.4)。s2中dna序列除了被甲基化以外,其他均与s1中的dna序列相同。
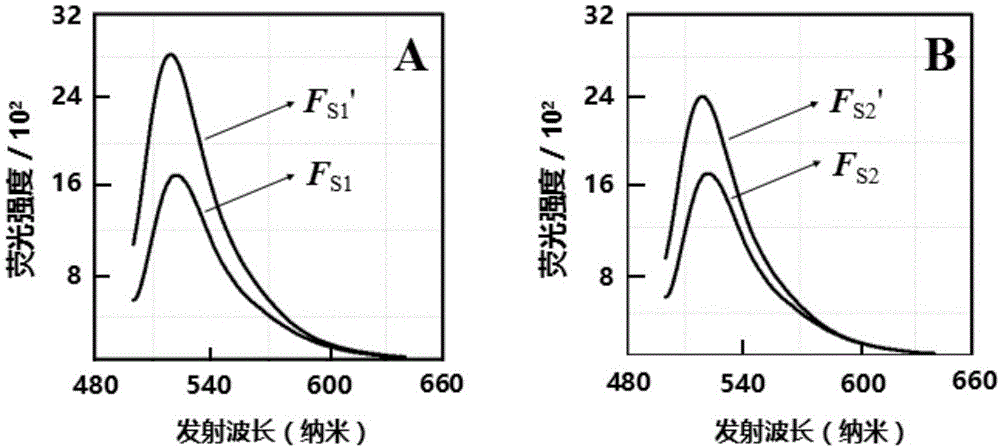
(3)分别抽取50μls1、s2至比色皿,在荧光仪中检测荧光信号(激发光ex:488nm,发射光em:500~640nm),分别记录荧光信号值为fs1、fs2。
(4)将s1和s2置于37℃的恒温环境中温浴2小时。分别抽取50μls1和s2至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm),分别记录荧光信号值为fs1’、fs2’。
(5)对比fs1和fs1’,fs2和fs2’,验证fret效果。对比fs1’和fs2’,验证甲基化dna对fret效果的影响。
检测结果如图2所示,fs1’和fs2’明显大于fs1和fs2,此现象表明dna探针链和互补连杂交后,形成了碱基间的π电子云堆积,促成了fret。同时,fs2’比fs1’低约16%,表明甲基化dna对fret具有一定的阻碍作用。
实施例2甲基化转移酶在不同温度下的作用状态
以dna甲基化转移酶dnmt3a作用甲基化位点5’-ccgg-3’为例。
(1)配制1ml样本储备液(s3),其含有500nm标记5-fam的dna杂交链(其中一条dna单链的荧光标记在甲基化位点向3’端方向的3bp以内的核苷酸上,另一互补序列的荧光标记在3’端,两处标记的荧光均为5-fam)、1×ne缓冲液(ph=7.4)、150nmdnmt3a、150μmsam;所述1×ne缓冲液中含有50mmnacl(氯化钠),10mmtris-hcl(三(羟甲基)氨基甲烷-盐酸溶液),10mmmgcl2(氯化镁)and1mmdtt(二硫苏糖醇)。。
(2)抽取50μls3至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm),记录初始信号值(fs3-0)。
(3)将s3置于37℃的恒温环境中15分钟,抽取50μls3至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm),记录信号值(fs3-1)。之后将s3置于49℃的恒温环境中5分钟,抽取50μls3至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm),记录信号值(fs3-1’)。
(4)重复步骤(3)四次,记录每次检测的信号值(fs3-2、fs3-2’、fs3-3、fs3-3’、fs3-4、fs3-4’、fs3-5、fs3-5’)。
(5)分别计算fs3-1...fs3-5、fs3-1’...fs3-5’与fs3-0的差值,结果用“荧光信号降低百分率”表示,即declinen=(fs3-0-fs3-n)/fs3-0×100%,declinen′=(fs3-0-fs3-n′)/fs3-0×100%,根据计算结果绘制信号变化曲线。
结果图如图3所示,初始时期,经49℃温浴处理后所产生的fret信号差异,与经37℃温浴处理后所产生的fret信号差异差距最大。随着循环数的增加,该差距逐渐减小。这主要是由于目标位点逐步被甲基化而引起的。初期甲基化点位未生成,在37℃时dnmt3a对目标dna的吸附作用最为明显,产生较大的非特异性fret信号差异。在49℃时,dnmt3a脱附于目标dna,此时fret信号干扰主要来源于甲基化位点,然而该阶段甲基化位点基本未出现,因此未产生明显信号差异。后期,随着甲基化位点的逐渐产生,dnmt3a对目标dna失去敏感度,其吸附作用逐渐减弱,产生的非特异性fret信号差异逐渐减小(decline2>decline3>decline4>decline5),同时,甲基化位点的增多使得特异性fret信号差异逐渐增大(decline2’<decline3’<decline4’<decline5’),形成了与dnmt3a吸附作用相反的信号差异趋势。当目标dna被完全甲基化后,37℃下,dnmt3a不再对其产生吸附作用,此时fret信号差异完全来自于甲基化位点。
实施例3甲基转移酶吸附时间的优化
(1)配制4份1ml样本储备液(s4、s5、s6、s7),每份含有500nm标记5-fam的dna杂交链(其中一条dna单链的荧光标记在甲基化位点向3’端方向的3bp以内的核苷酸上,另一互补序列的荧光标记在3’端,两处标记的荧光均为5-fam)、1×ne缓冲液(ph=7.4)、150nmdnmt3a、150μmsam。所述1×ne缓冲液中含有50mmnacl(氯化钠),10mmtris-hcl(三(羟甲基)氨基甲烷-盐酸溶液),10mmmgcl2(氯化镁)and1mmdtt(二硫苏糖醇)。
(2)分别抽取50μls4、s5、s6、s7至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm),记录初始荧光信号值(fs4-0、fs5-0、fs6-0、fs7-0)。
(3)分别将s4、s5、s6、s7置于37℃的恒温环境中3、5、7、9分钟,再将其置于49℃的恒温环境中,固定为3分钟,抽取50μl至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm)。之后重复上述过程,直至反应总时长达100分钟,记录各组荧光信号值(fs4、fs5、fs6、fs7)。
(4)分别计算fs4、fs5、fs6、fs7与fs4-0、fs5-0、fs6-0、fs7-0的差值,结果用“荧光信号强度归一化比率”表示。根据计算结果绘制信号变化曲线,确定dnmt3a最佳吸附时间。
图4中显示,当甲基转移酶的吸附时间设置为3分钟时,fret信号的整体降低率偏低(图4中的曲线a),表明目标dna的甲基化效果相对不明显。随着甲基转移酶的吸附时间的增长,fret信号的整体降低率逐渐增大,表明目标dna的甲基化效果逐渐显现。当甲基转移酶吸附时间达到7分钟时,fret信号的整体降低率达到平台期(图4中的曲线c)。因此,甲基转移酶吸附时间为7分钟时较好。
实施例4甲基转移酶脱附时间的优化
(1)配制4份1ml样本储备液(s8、s9、s10、s11),每份含有500nm标记5-fam的dna杂交链(其中一条dna单链的荧光标记在甲基化位点向3’端方向的3bp以内的核苷酸上,另一互补序列的荧光标记在3’端,两处标记的荧光均为5-fam)、1×ne缓冲液(ph=7.4)、150nmdnmt3a、150μmsam。所述1×ne缓冲液中含有50mmnacl(氯化钠),10mmtris-hcl(三(羟甲基)氨基甲烷-盐酸溶液),10mmmgcl2(氯化镁)and1mmdtt(二硫苏糖醇)。
(2)分别抽取50μls8、s9、s10、s11至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm),记录初始信号值(fs8-0、fs9-0、fs10-0、fs11-0)。
(3)分别将s8、s9、s10、s11置于37℃的恒温环境中,固定为7分钟,再将其置于49℃的恒温环境中1、3、5、7分钟,抽取50μl至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm)。之后重复上述过程,直至反应总时长达100分钟,记录各组荧光信号值(fs8、fs9、fs10、fs11)。
(4)分别计算别计算fs8、fs9、fs10、fs11与fs8-0、fs9-0、fs10-0、fs11-0的差值,结果用“荧光信号强度归一化比率”表示。根据计算结果绘制信号变化曲线,确定甲基转移酶最佳脱附时间。
结果如图5所示,当甲基转移酶的脱附时间被设置为1分钟(图5中的曲线d),fret信号的整体降低率偏大,表明甲基转移酶吸附对fret信号有影响;而随着甲基转移酶的脱附时间的增长,fret信号的整体降低率逐渐减小,表明甲基转移酶脱附于目标dna的效果逐渐显现。但当甲基转移酶脱附时间达到7分钟时(图5中的曲线a),fret信号的整体降低率偏小,表明该脱附时间下,甲基转移酶的作用效能受到影响。以fret信号的整体降低率达到适中值(即甲基化dna对fret的影响值:16%)为标准,甲基转移酶脱附时间为3min、5min时均满足要求,其中最佳脱附时间确定为3分钟(图5中的曲线b)。
实施例5dna甲基化动态过程的高通量在线检测方法
(1)配制1ml样本储备液(s13),其含有500nm标记5-fam的dna杂交链(其中一条dna单链的荧光标记在甲基化位点向3’端方向的3bp以内的核苷酸上,另一互补序列的荧光标记在3’端,两处标记的荧光均为5-fam)、1×ne缓冲液(ph=7.4)、150nmdnmt3a、150μmsam。所述1×ne缓冲液中含有50mmnacl(氯化钠),10mmtris-hcl(三(羟甲基)氨基甲烷-盐酸溶液),10mmmgcl2(氯化镁)and1mmdtt(二硫苏糖醇)。
(2)抽取30μls13至pcr样管中,在定量pcr仪中设置参数:37℃、7分钟,49℃、3分钟;共计12个循环;进行在线测试,记录结果数据。
或者,抽取30份s13(每份30μl)至96孔pcr样板中,设置定量pcr仪运行参数:37℃、7分钟,49℃、3分钟;共计12个循环;进行在线测试,记录结果数据。
图6结果表明,无论单一样本(a),还是多组分样本(b),fret信号差异都会随着时间增加而逐渐增大,并最终达到平台期(80~120分钟)。由于排除了dnmt3a吸附的影响,此信号差异来源于甲基化dna的生成。测试结果显示,甲基化dna对fret的整体降低值约17.27%,对比前期测定的标准影响值(16%),定量pcr测试结果拥有较高的吻合度(92.07%)。另外,需要补充说明的,图6是pcr仪对30份样本同时检测,但其对每一份样本的光激发值都有偏差。其最大光源点位在96孔板的中心,光源强度随圆心半径增大而减小,结果体现在每一份样本的初始光强都不一样(中心样本最大,边缘样本相对减弱),但每一份样本fret信号差异都会随着时间增加而逐渐增大,并最终达到平台期(80~120分钟)。
对比例1dna甲基化过程离线检测
(1)配置1ml样本储备液(s12),其含有500nm标记5-fam的dna杂交链(其中一条dna单链的荧光标记在甲基化位点向3’端方向的3bp以内的核苷酸上,另一互补序列的荧光标记在3’端,两处标记的荧光均为5-fam)、1×ne缓冲液(ph=7.4)、150nmdnmt3a、150μmsam。所述1×ne缓冲液中含有50mmnacl(氯化钠),10mmtris-hcl(三(羟甲基)氨基甲烷-盐酸溶液),10mmmgcl2(氯化镁)and1mmdtt(二硫苏糖醇)。
(2)抽取50μls12至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm),记录初始信号值(fs12-0)。
(3)将s12置于37℃的恒温环境中7分钟,再将其置于49℃的恒温环境中3分钟,抽取50μls12至比色皿,在荧光仪中检测荧光信号(ex:488nm,em:500~640nm)。之后重复上述过程直至反应总时长达100分钟,记录每次检测的荧光信号值(fs12)。
(4)分别计算fs12与fs12-0的差值,结果用“荧光信号强度归一化比率”表示。根据计算结果绘制信号变化曲线。
图7中显示,随着时间增加,fret信号差异逐渐增大,由于排除了dnmt3a吸附的影响,此信号差异来源于甲基化dna的生成。反应过程中,前15分钟为dna甲基化的起始期,15~75分钟为dna甲基化的生成期,75~100分钟为dna甲基化的稳定期。测试结果显示,甲基化dna对fret的整体降低值约18.79%,与前期测定(图2所示)的标准影响值(16%)较吻合。当目标dna趋于完全甲基化(75~100分钟)时,dnmt3a会处于游离与吸附的不稳定状态,容易受到测试环境的干扰。但在在线检测中,这种现象会得到有效改善。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!