单电子转移活性自由基聚合制备星形聚丙烯酰胺的方法与流程
本发明涉及丙烯酰胺类聚合物制备领域,具体涉及单电子转移活性自由基聚合制备星形聚丙烯酰胺的方法。
背景技术:
1948年flory首次提出星型聚合物这一概念。星形聚合物具有紧凑的三维核壳空间结构,分子内和分子间不易发生交联,多臂上的极性官能团高度集中、模量高,这使得星形聚合物具有一些特殊的性质,如结晶度低、扩散系数、熔融粘度、流体动力学体积较小等因此被制成一系列可用于药物递送,凝集素测定,癌症治疗以及光子学等领域的纳米材料。
聚丙烯酰胺(pam)是丙烯酰胺单体均聚或者与其他单体进行共聚得到聚合物的统称。在丙烯酰胺分子结构中含有酰胺基易形成氢键,使其具有优良的水溶性。分子量大小在很大程度上决定着产品的用途及功能,高分子量的聚丙烯酰胺(105~107)对许多固体表面和溶解物质有着良好的粘附力,因而应用于增稠、絮凝、阻垢、采油及生物医学材料等领域;中等分子量的可用作造纸行业的纸张干燥剂;低分子量的则用作油墨分散剂。聚丙烯酰胺产品在工业领域的应用呈稳步上升的趋势。
目前合成聚丙烯酰胺的常用方法是传统的水溶液自由基聚合。虽然该方法聚合工序简单、成本较低,但生产过程中单体转化率较低,聚合的产物固含量仅在8%~25%,且容易发生酰亚胺化反应,生成凝胶,此外传统自由基聚合法得到的产物结构和分子量不可控,分子量分布较宽。
技术实现要素:
本发明的一个目的是提供单电子转移活性自由基聚合制备星形聚丙烯酰胺的方法,这种单电子转移活性自由基聚合制备星形聚丙烯酰胺的方法用于解决传统的水溶液自由基聚合制备聚丙烯酰胺的方法可控性不强的问题。
本发明解决其技术问题所采用的技术方案是:这种单电子转移活性自由基聚合制备星形聚丙烯酰胺的方法:
步骤一、聚氧乙基甘油醚与2-溴代异丁酰溴反应制备水溶性三臂引发剂gly-br3;
步骤二、在反应瓶
上述方案中步骤二中歧化过程的气氛条件为在惰性气体条件下,歧化时间为2min~30min;反应瓶
上述方案中步骤二中催化剂为cubr与配体原位歧化生成的cu0。
上述方案中单体为丙烯酰胺或丙烯酰胺、n-乙烯基吡咯烷酮(nvp)、2-丙烯酰胺基-2-甲基丙磺酸钠(naamps)或丙烯酰胺、4-乙烯基吡啶(4vp)或丙烯酰胺乙烯基单体。
上述方案中乙烯基单体为n-烷基丙烯酰胺或丙烯酸钠或甲基丙烯酸二甲氨基乙酯。
上述方案中的惰性气体为氮气或氩气。
上述方案中含氮多齿化合物为三(n,n'-二甲基氨基乙基)胺(me6-tren)、n-丙基-2-吡啶基-甲胺(pr-pmi)、n,n,n',n'-四甲基乙二胺(tmeda)、1,4,8,11-四氮环十四烷(cyclam)、三胺基乙基胺(tren)、五甲基二乙烯三胺(pmdeta)、六甲基三亚乙基四胺(hmteta)中任意一个。
本发明具有以下有益效果:
1、本发明采用活性自由基的最新研究方法单电子转移活性自由基聚合法(set-lrp),在室温(或低于室温度)的条件下,以绿色溶剂水为反应介质,制备出星形聚丙烯酰胺;本发明聚合速率快,单体转化率高(10min内可达95%),链端活性链端保留率高,分子量可控,分子量分布较窄(最低可达1.18)。
2、本发明节能环保,操作简便,为星形聚丙烯酰胺的工业化生产提供便利。
3、为了获得理想的聚合反应条件,本发明以聚氧乙烯甘油醚为原料合成水溶性三臂星形引发剂,并首次以此为核引发剂引发丙烯酰胺单体的均聚及共聚。
4、本发明所用催化剂通过cuix/l原位歧化制备,歧化温度决定生成的cu0纳米粒子的尺寸,可通过调整温度来控制cu0的大小(即cu0的表面积),从而达到控制聚合反应速率及所得聚合物产品的性能。
5、水代替传统有机溶剂作为聚合反应介质,不仅具有成本低廉、环境友好、价格低廉、适合用作生物大分子合成介质的优势,而且具有溶剂化作用强、加速cuix/l歧化、加快丙烯酰胺类单体聚合动力学的作用。
6、本发明合成路线,多种乙烯基结构的单体均可以与丙烯酰胺单体进行无规共聚,甚至可以与其发生嵌段聚合,极大地丰富了星形丙烯酰胺类聚合物的种类。
附图说明
图1为实施例1中三臂星形pam的1hnmr;
图2为实施例1中三臂星形pam的1cnmr;
图3为实施例1中聚合反应动力学曲线;
图4为实施例2中聚合反应动力学曲线;
图5为实施例2中聚合物分子量和分子量分布与理论分子量关系曲线;
图6为实施例3中聚合物的ft-ir谱图;
图7为实施例3中聚合物的1hnmr谱图;
图8为实施例4中聚合物的1hnmr谱图。
具体实施方式
下面结合附图对本发明作进一步的说明:
实施例1:
室温条件下,在带有磁子标号为
图1为三臂星形pam的1hnmr谱图,产物主链上的质子峰h(c,d)出现在1.34-1.63ppm;h(b)受到cl端基和酰胺基的共同影响,质子峰出现在3.50-3.32ppm;h(e)受到醚键的影响,质子峰出现在1.78-2.41;h(a)的质子峰没有出现在1hnmr图中,这是由于-nh2氢非常活跃,可以与溶剂质子交换。而4.63ppm附近出现的强吸收峰为溶剂峰。
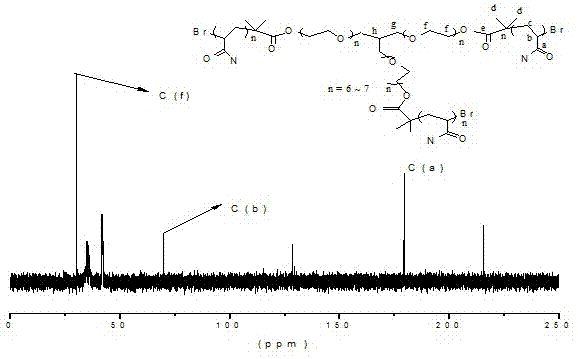
图2为三臂星形pam的1cnmr谱图,聚合物主链上c(f)峰出现在30.15ppm附近,受酰胺基影响c(a)峰出现在179.36ppm附近,c(b)受br端基影响峰出现在70ppm附近,结合图1及图2可确定得到的产物为三臂星型pam。
图3为该聚合反应动力学曲线图,从图中可以看出,ln([m]0/[m])随时间呈线性增加,这表明一级增长速率与自由基和单体浓度有关,自由基浓度在聚合过程中为常数,链增长速率常数
(转化率在15min可达到95%,mngpc=41500)
实施例2:
参照实施例一的配比进行am的set-lrp聚合,不同的是聚合反应在0℃条件下进行。
重量法测得单体转化率为100%,gpc测得聚合物mngpc=38900,mw/mn=1.19
图4为该聚合反应动力学曲线图,与实施例一图3相比,聚合反应温度从室温(25℃)降低到0℃,链增长速率常数
图5为该反应聚合物分子量和分子量分布与理论分子量关系曲线图,从图中可以看出,实际分子量与理论分子量相近,分子量分布随反应的进行而减小,这都符合活性可控聚合的规律。
实施例3:
在带有磁子标号为i的反应瓶中,加入去离子h2o(2ml)与配体(me6-tren,10.7µl,0.04mmol)通入氮气10分钟后加入cubr(0.0057g,0.04mmol),继续通氮气使cubr在无氧条件下歧化0.5h。同时将单体(am,0.7463g,10.5mmol;nvp,0.0333g,0.3mmol;2-丙烯酰胺基-2-甲基丙磺酸钠(naamps),0.15ml,0.3mmol),引发剂gly-br3(0.0211g,0.0111mmol)以及一定量去离子h2o共3ml加入标号为ii的反应瓶中,并使其混合均匀,通入氮气10min。待歧化完成后,将反应瓶ii中溶液用注射器注入反应瓶i中继续通氮气,并在磁力搅拌机上搅拌,一定时间后取出,并用过量丙酮沉淀出聚合物,经中性al2o3柱层析除去未反应的cu0粉及二价铜与配体的络合物,将得到的澄清聚合物溶液用过量丙酮沉淀,真空干燥至恒重,即得目标产物。
重量法测得单体转化率为92%,gpc测得聚合物mngpc=8.21×104,mw/mn=1.23
图6为星形聚合物p(am-naamps-nvp)的ftir。ir(kbr):3182cm-1是酰胺基n-h的特征吸收峰;2927cm-1和2791cm-1分别vasc-h和vsc-h的特征双峰;1319cm-1是nvp结构中-c=o伸缩振动峰;1449cm-1是nvp结构中酰胺基团c-n键的伸缩振动峰;1116cm-1和1040cm-1是磺酸基团的特征吸收峰;产物红外光谱中同时存在着am、naamps、nvp的特征吸收峰,初步证明所得产物确实为三种单体的共聚物。
图7为星形聚合物p(am-naamps-nvp)的1hnmr谱图。1h-nmr(400mhz,d2o,δ):7.7ppm为h(a)的质子峰;6.8ppm为h(b)的质子峰;δ=1.78ppm为h(i)的质子峰;δ=2.2ppm为h(g)的质子峰;δ=5.8ppm为h(k)的质子峰。结合图5,可以确定该实验条件下得到的聚合物为目标产物,同时聚合物链端含有α-br端基,也表明了聚合反应为活性聚合,按set-lrp聚合机理进行。
实施例4:
冰水浴中,在带有磁子标号为i的反应瓶中,加入去离子h2o(2ml)与配体(me6-tren,10.6µl,0.04mmol)通入氮气10分钟后加入cubr(0.0086g,0.06mmol),继续通氮气使cubr在无氧条件下歧化0.5h。与此同时,将单体(am,0.5120g,7.2mmol),引发剂gly-br3(0.1200g,0.06mmol)以及一定量去离子h2o共3ml加入标号为ii的反应瓶中,并使其混合均匀,通入氮气10min;标号为iii的反应瓶中加入4-乙烯基吡啶(4vp,0.7570g,7.2mmol),2ml去离子水,通入氮气;i号反应瓶歧化完成后用注射器将ii号反应瓶中的物质加入其中,搅拌,1hnrm监测am转化率,待am反应完全,用注射器将iii号反应瓶中的物质加入i号瓶,聚合反应结束后将产物过滤、透析,透析产品在-50℃下真空冻干,即得目标产物p(am-b-4vp)。
图8为三臂星形嵌段聚合物p(am-b-4vp)核磁共振氢谱图。对比图1三臂星形均聚pam的1hnmr可以看出,pam成功保留链端活性基团并与4vp单体进行嵌段共聚,得到目标嵌段聚合物p(am-b-4vp)。
以上实施例仅通过丙烯酰胺与n-乙烯基吡咯烷酮(nvp)、2-丙烯酰胺基-2-甲基丙磺酸钠(naamps)及4-乙烯基吡啶(4vp)单体进行共聚作为实施例,其他乙烯基单体如n-烷基丙烯酰胺、丙烯酸钠、甲基丙烯酸二甲氨基乙酯等亦可与丙烯酰胺单体共聚,均适用本发明的技术路线。
对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,在不脱离本发明范围的情况下,在其它实施例中实现,因此,本发明将不会被限制于本文所示的这些实施例。
- 还没有人留言评论。精彩留言会获得点赞!