一种高对映选择性2,3-二取代含硼吲哚类化合物及其制备方法与流程
本发明属于有机合成化学技术领域,具体涉及一种高对映选择性2,3-二取代含硼吲哚类化合物及其制备方法。
背景技术:
具有光学活性的2,3-二取代吲哚在有机合成和药物化学中具有非常广泛的应用,在众多的天然产物和药物活性分子中都存在这种杂环结构。此外,这类手性杂环骨架也是药物发现的关键基础,很多类天然分子库都是以吲哚结构框架为基础的。因此,氮杂环结构的不对称合成策略相继已被研究报道。在这些方法中,酸催化的吲哚的不对称氢化或许是最重要的方法。尽管该方法极具吸引力,但是由于需要提供强酸性环境、高压条件以及较差的官能团兼容性限制了其广泛的应用。其他值得注意的方法,例如吲哚的动力学拆分、钯催化的不对称分子内偶联反应、串联/芳香族亲核取代反应(tandem AN/SNAr processes),不对称的sparteinemediated反应也有所发展,但是苛刻的反应条件、多步合成以及较低的产率,限制了其合成应用。更为重要的是,利用以上方法得到的吲哚类化合物没有进行后续修饰的潜力。因此在一步合成吲哚类化合物的同时引入一个能进行后续修饰的含硼官能团,能极大地扩展这种具有重要生理活性的手性吲哚类化合物的种类和数量,对药物的进一步修饰和改造具有重要意义。但是,迄今,还没有直接的不对称方法来得到手性2,3-二取代的硼化吲哚类化合物。尤其在药用杂环化合物(例如,吲哚类、喹啉类、吡啶类)和吲哚的不对称塑造尚未熟知。
随着全球生态环境的急剧恶化,如何实现可持续发展已成为人类面临的重大问题,以从源头上消除污染、节省资源为核心的绿色化学研究已经成为解决日益严峻的生态环境问题的强有力手段。Cu催化邻亚胺基苯乙烯衍生物的分子内不对称硼碳化反应就具有环境友好、价格低廉、实用高效等优点,可以同时构建两个相邻位置的手性中心。到目前为止,具有高对映选择性硼化的2,3-二取代吲哚类化合物的反应还未见文献报道。
技术实现要素:
本发明的目的是提供一种高对映选择性2,3-二取代含硼吲哚类化合物及其制备方法,该化合物具有高对映选择性。
本发明首先提供一种高对映选择性2,3-二取代含硼吲哚类化合物,该化合物的结构通式如式Ⅰ所示:
式Ⅰ中,R1为氢、烷基、烷氧基或卤素,R2为烷基、带有给电子基的芳基、带有吸电子基的芳基以及杂芳基,-Bpin代表
优选的是,所述的R1为氢、-CH3、-F。
优选的是,所述的R2为
优选的是,所述的高对映选择性2,3-二取代含硼吲哚类化合物,具有2a-2l所示的结构:
本发明还提供一种高对映选择性2,3-二取代含硼吲哚类化合物的制备方法,该方法包括:
氮气条件下,在反应容器中加入催化剂、配体(S,S)-Ph-BPE、碱和溶剂,室温下搅拌,然后加入联硼酸频那醇酯继续搅拌,再加入官能化的邻亚氨基苯乙烯,室温反应,得到2,3-二取代含硼吲哚类化合物。
优选的是,所述的催化剂为氯化亚铜。
优选的是,所述的碱为NaOtBu。
优选的是,所述的溶剂为甲苯、乙醚、四氢呋喃、甲基叔丁基醚或二氯甲烷。
优选的是,所述的反应时间为6-8h。
优选的是,所述的催化剂、配体(S,S)-Ph-BPE、碱、联硼酸频那醇酯和官能化的邻亚氨基苯乙烯的摩尔比为2:3:0.3:0.3:0.2。
本发明的有益效果
本发明首先提供一种高对映选择性2,3-二取代含硼吲哚类化合物,该化合物的结构通式如式Ⅰ所示。本发明的化合物是通过氯化亚铜催化剂和手性配体(S,S)-Ph-BPE产生手性铜配合物,该手性铜在碱(tBuONa)的作用下生成手性的叔丁氧铜活性催化剂,之后与联硼酸频哪醇酯生成铜硼物种插入碳碳双键,随后对亚胺官能团进一步环合,最后在醇的作用下得到分子内硼碳化的产物。本发明的2,3-二取代吲哚类化合物具有高对映选择性,通过高效液相色谱实验证明其ee值最高可达99%。
本发明还提供一种高对映选择性2,3-二取代含硼吲哚类化合物的制备方法,该方法操作简单,原料试剂易得,条件温和,反应体系绿色环保,产物易分离纯化,适用于合成多种高对映选择性的2,3-二取代吲哚类化合物,其中,3-位烷基硼取代基可以进一步衍生化,将硼基团转化成羟基、氨基、多一个碳的硼基团或羟基等。另外,经克级实验证明,本发明可适用于大规模工业生产,可以高效、高效率地制得高纯度的2,3-二取代吲哚类化合物。
附图说明
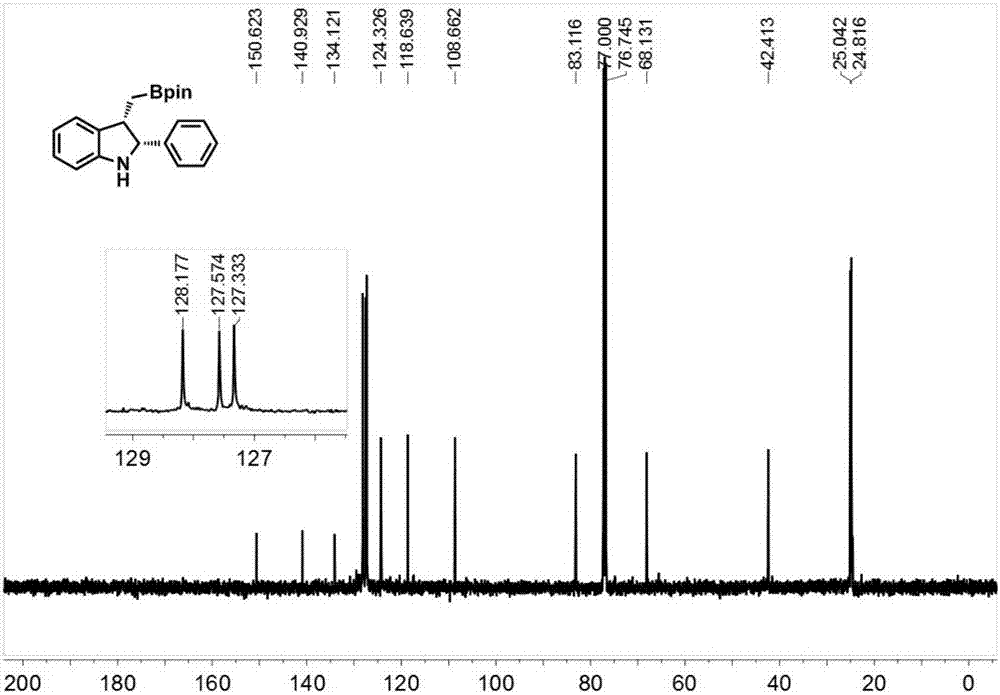
图1为实施例1制备得到的2,3-二取代吲哚2a的1H-NMR的核磁共振谱;
图2为实施例1制备得到的2,3-二取代吲哚2a的13C-NMR的核磁共振谱;
图3为本发明实施例1外消旋产物的高效液相色谱图;
图4为本发明实施例1制备得到的2,3-二取代吲哚2a的高效液相色谱图;
图5为本发明实施例1制备得到的2,3-二取代吲哚2a重结晶后的高效液相色谱图;
图6为本发明实施例3外消旋产物的高效液相色谱图;
图7为本发明实施例3制备得到的2,3-二取代吲哚2c的高效液相色谱图;
图8为本发明实施例6外消旋产物的高效液相色谱图;
图9为本发明实施例6制备得到的2,3-二取代吲哚2f的高效液相色谱图;
图10为本发明实施例8外消旋产物的高效液相色谱图;
图11为本发明实施例8制备得到的2,3-二取代吲哚2h的高效液相色谱图;
图12为实施例11制备得到的2,3-二取代吲哚2k的1H-NMR的核磁共振谱;
图13为实施例11制备得到的2,3-二取代吲哚2k的13C-NMR的核磁共振谱;
图14为本发明实施例11外消旋产物的高效液相色谱图;
图15为本发明实施例11制备得到的2,3-二取代吲哚2k的高效液相色谱图。
具体实施方式
本发明首先提供一种高对映选择性2,3-二取代含硼吲哚类化合物,该化合物的结构通式如式Ⅰ所示:
式Ⅰ中,R1为氢、烷基、烷氧基或卤素,优选为氢、-CH3或-F;R2为烷基、带有给电子基的芳基、带有吸电子基的芳基以及杂芳基,更优选为-Bpin代表
按照本发明,所述的高对映选择性2,3-二取代含硼吲哚类化合物,具有2a-2l所示的结构:
本发明还提供一种高对映选择性2,3-二取代含硼吲哚类化合物的制备方法,该方法包括:
氮气保护下,在反应容器中加入催化剂、配体(S,S)-Ph-BPE、碱和溶剂,室温下搅拌,所述的搅拌时间优选为8-15min,更优选为10min,然后加入联硼酸频那醇酯继续搅拌,所述的搅拌时间优选为8-15min,更优选为10min,再加入官能化的邻亚氨基苯乙烯,优选先搅拌5min后,然后加入甲醇室温反应,所述的反应时间为6-8h,并用TLC检测反应完成,然后就经萃取,合并有机相,用无水硫酸钠干燥、抽滤,减圧蒸馏除去有机溶剂,最后经过硅胶柱层析,得到2,3-二取代含硼吲哚类化合物。
按照本发明,所述的催化剂优选为氯化亚铜;所述的碱优选为NaOtBu;所述的溶剂优选为甲苯、乙醚、四氢呋喃、甲基叔丁基醚或二氯甲烷;所述的催化剂、配体(S,S)-Ph-BPE、碱、联硼酸频那醇酯和官能化的邻亚氨基苯乙烯的摩尔比优选为2:3:0.3:0.3:0.2。
所述的制备方法的具体反应过程如下:
按照本发明,所述的官能化的邻亚氨基苯乙烯按照现有技术的已知方法制备而成,具体可参考文献Ascic,E.;Buchwald,S.L.J.Am.Chem.Soc.2015,137,4666-4669。
按照本发明,本发明的方法是氯化亚铜催化剂和手性配体(S,S)-Ph-BPE产生手性铜配合物,该手性铜在碱(tBuONa)的作用下生成手性的叔丁氧铜活性催化剂,之后与联硼酸频哪醇酯生成铜硼物种插入碳碳双键,随后对亚胺官能团进一步环合,最后在醇的作用下得到分子内硼碳化的产物。本发明的2,3-二取代吲哚类化合物具有高对映选择性,通过高效液相色谱实验证明其ee值最高可达99%。
下面结合具体实施例对本发明做进一步详细的说明,实施例中涉及到的原料均为商购获得。
实施例1
氮气保护下,向带有搅拌子的10mL耐压封管中加入氯化亚铜(2mol%)、(S,S)-Ph-BPE(3mol%)和NaOtBu(0.30mmol),再加入四氢呋喃(2mL),室温下搅拌10min。搅拌10min后,向体系中加入联硼酸频哪醇酯(0.30mmol),继续搅拌10min,然后,加入1a(0.20mmol),搅拌5min后加入甲醇(0.20mmol)将反应放在室温搅拌8h,并用TLC检测反应完成。反应用水(10ml)淬灭,二氯甲烷(3×10mL)萃取,合并有机相,用无水硫酸钠干燥、抽滤,减圧蒸馏除去有机溶剂,最后经过硅胶柱层析,得到2,3-二取代吲哚类化合物2a,产率为96%。
图1为实施例1制备得到的2,3-二取代吲哚2a的1H-NMR的核磁共振谱;图2为实施例1制备得到的2,3-二取代吲哚2a的13C-NMR的核磁共振谱;谱图解析数据2a:
White solid.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.30–7.22(m,5H),7.19(d,J=7.0Hz,1H),7.04(t,J=7.5Hz,1H),6.72(t,J=7.5Hz,1H),6.65(d,J=8.0Hz,1H),4.95(d,J=8.0Hz,1H),4.03(s,1H),3.69(q,J12=8.5Hz,J13=15.5Hz,1H),1.20(d,J=3.5Hz,12H),0.90–0.85(m,1H),0.56–0.52(m,1H);13C NMR(125MHz,CDCl3)δ=150.62,140.93,134.12,128.18,127.57,127.33,124.33,118.64,108.66,83.12,68.13.42.41,25.04,24.82.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for Calcd for C21H27BNO2([M+H]+),336.2134,found,336.2130.[α]D25=+4.9,(c=1,CHCl3).HPLC analysis(AD-H,5%IPA/hexane,1mL/min,250nm)indicated 90%ee:tR(major)=7.7min,tR(minor)=12.9min.
图3为本发明实施例1外消旋产物的高效液相色谱图,通过图3的外消旋产物的结构确定对照保留时间,然后得到实施例1制备产物的高效液相色谱图,如图4所示,ee值为92%。将实施例1得到的产物进行重结晶,得到的高效液相色谱图如图5所示。
实施例2
用1b代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2b,ee值为98%。。
实施例2制备得到的产物2b的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
White solid.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.17(d,J=8.0Hz,3H),7.07(d,J=8.0Hz,2H),7.04(d,J=7.5Hz,1H),6.72(t,J=7.5Hz,1H),6.66(d,J=7.5Hz,1H),4.93(d,J=8.5Hz,1H),4.01(s,1H),3.66(q,J12=8.5Hz,J13=15.5Hz,1H),2.31(s,3H),1.20(d,J=5.0Hz,12H),0.90–0.85(m,1H),0.58–0.54(m,1H);13C NMR(125MHz,CDCl3)δ=150.66,137.82,136.88,134.19,128.82,127.46,127.26,124.28,118.53,108.60,83.60,67.87,42.35,25.02,24.80,21.07.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C22H29BNO2([M+H]+),350.2290,found,350.2290.[α]D19=+15.8,(c=1,CHCl3).HPLC analysis(AD-H,5%IPA/hexane,1mL/min,250nm)indicated 84%ee.After recrystallization,the ee value of the filtrate was 98.4%:tR(major)=6.2min,tR(minor)=13.2min.
实施例3
用1c代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2c。
实施例3制备得到的产物2c的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
White solid.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.19(t,J=8.0Hz,3H),7.04(t,J=7.5Hz,1H),6.80(d,J=7.5Hz,2H),6.72(t,J=7.0Hz,1H),6.65(d,J=7.5Hz,1H),4.92(d,J=8.0Hz,1H),4.01(s,1H),3.77(s,3H),3.64(q,J12=7.5Hz,J13=14.5Hz,1H),1.21(d,J=3.0Hz,12H),0.91–0.86(m,1H),0.59–0.54(m,1H);13C NMR(125MHz,CDCl3)δ=158.79,150.59,134.14,132.93,128.60,127.25,124.27,118.53,113.45,108.58,83.06,67.54,55.18,42.37,25.03,24.79.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C22H29BNO3([M+H]+),366.2239,found 366.2233.[α]D19=+14.9,(c=1,CHCl3).HPLC analysis(AD-H,10%IPA/hexane,1mL/min,250nm)indicated 96.7%ee:tR(major)=7.1min,tR(minor)=14.9min.
图6为本发明实施例3外消旋产物的高效液相色谱图,通过图6的外消旋产物的结构确定对照保留时间,然后得到实施例3制备产物的高效液相色谱图,如图7所示,ee值为96%。
实施例4
用1d代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2d,ee值为89%。
实施例4制备得到的产物2d的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
White solid.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.29(d,J=8.0Hz,2H),7.22(d,J=8.0Hz,2H),7.19(d,J=7.5Hz,1H),7.05(t,J=7.5Hz,1H),6.72(t,J=7.5Hz,1H),6.66(d,J=8.0Hz,1H),4.94(d,J=8.5Hz,1H),4.02(s,1H),3.71–3.66(m,1H),1.29(s,9H),1.20(d,J=5.0Hz,12H),0.89–0.84(m,1H),0.65–0.61(m,1H);13C NMR(125MHz,CDCl3)δ=150.67,150.15,137.82,134.29,127.36,127.28,125.05,124.32,118.57,108.62,83.09,67.89,42.33,34.44,31.35,25.06,24.81.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C25H35BNO2([M+H]+),392.2760,found,392.2760.[α]D19=+12.6,(c=0.5,CHCl3).HPLC analysis(AD-H,Hexane/IPA=95/5,1mL/min,250nm)indicated 89%ee:tR(major)=5.0min,tR(minor)=13.1min.
实施例5
用1e代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2e,ee值为79%。
实施例5制备得到的产物2e的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
Colorless oil.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.54(d,J=8.0Hz,2H),7.45(d,J=8.0Hz,2H),7.21(d,J=7.0Hz,1H),7.07(t,J=7.5Hz,1H),6.75(t,J=7.5Hz,1H),6.69(d,J=8.0Hz,1H),5.03(d,J=8.0Hz,1H),4.07(s,1H),3.72(q,J12=8.5Hz,J13=15.0Hz,1H),1.19(d,J=2.5Hz,12H),0.87–0.82(m,1H),0.52–0.47(m,1H).13C NMR(125MHz,CDCl3)δ=150.15,145.01,133.87,129.53(q,J=32.1Hz),128.81,127.95,127.56,125.13(q,J=3.6Hz),124.45,124.15(q,J=270.5Hz),119.03,108.94,83.24,67.64,42.42,25.01,24.79.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C22H26BF3NO2([M+H]+),404.2007,found 404.2002.[α]D19=+4.9,(c=1,CHCl3).HPLC analysis(OD-H,5%IPA/hexane,1mL/min,250nm)indicated 72%ee:tR(major)=5.4min,tR(minor)=11.6min.
实施例6
用1f代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2f。
实施例6制备得到的产物2f的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
White solid.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.95(d,J=9.6Hz,2H),7.39(d,J=9.6Hz,2H),7.20(d,J=8.4Hz,1H),7.07(t,J=9.0Hz,1H),6.75(t,J=9.0Hz,1H),6.69(d,J=9.0Hz,1H),5.03(d,J=9.6Hz,1H),4.10(s,1H),3.90(s,3H),3.74–3.69(m,1H),1.20(d,J=3.6Hz,12H),0.89–0.83(m,1H),0.47–0.43(m,1H).13C NMR(125MHz,CDCl3)δ=166.93,150.25,146.31,133.82,129.51,129.14,127.54,127.50,124.39,118.88,108.86,83.19,67.78,52.03,42.46,25.01,24.79.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C23H28BNNaO4([M+Na]+),416.2008,found 416.1988.[α]D19=+5.8,(c=1,CHCl3).HPLC analysis(OD-H,10%IPA/hexane,1mL/min,250nm)indicated 90%ee:tR(major)=19.6min,tR(minor)=9.8min.
图8为本发明实施例6外消旋产物的高效液相色谱图,通过图8的外消旋产物的结构确定对照保留时间,然后得到实施例6制备产物的高效液相色谱图,如图9所示,ee值为90%。
实施例7
用1g代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2g,ee值为83%。
实施例7制备得到的产物2g的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
Colorless oil.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.40(d,J=8.5Hz,2H),7.20(d,J=8.5Hz,3H),7.06(t,J=8.0Hz,1H),6.75(t,J=7.5Hz,1H),6.69(d,J=7.5Hz,1H),4.94(d,J=8.0Hz,1H),3.67(q,J12=8.0Hz,J13=15.0Hz,1H),1.20(s,12H),0.88–0.83(m,1H),0.55–0.50(m,1H);13C NMR(125MHz,CDCl3)δ=139.79,134.04,131.28,129.33,127.50,124,42,121.13,119.05,108.99,83.24,67.48,42.31,25.03,24.81.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C21H26BBrNO2([M+H]+),414.1238,found 414.1225.[α]D19=+9.5,(c=0.2,CHCl3).HPLC analysis(AD-H,5%IPA/hexane,1mL/min,250nm)indicated 83%ee:tR(major)=6.6min,tR(minor)=13.2min.
实施例8
用1h代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2h。
实施例8制备得到的产物2h的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
White solid.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.17(d,J=7.5Hz,1H),7.09(t,J=3.5Hz,1H),7.07(t,J=7.5Hz,1H),6.91(d,J=3.5Hz,2H),6.77(t,J=7.5Hz,1H),6.68(d,J=8.0Hz,1H),5.22(d,J=8.0Hz,1H),4.15(s,1H),3.69(q,J12=7.5Hz,J13=15.5Hz,1H),1.24(d,J=5.0Hz,12H),1.09–1.04(m,1H),0.77–0.73(m,1H).13C NMR(125MHz,CDCl3)δ=149.74,144.46,133.46,127.36,126.23,125.17,124.19,123.97,119.18,109.30,83.18,64.32,42.75,25.02,24.85.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C19H25BNO2S([M+H]+),342.1697,found 342.1686.[α]D19=-14.8,(c=0.5,CHCl3).HPLC analysis(AD-H,5%IPA/hexane,1mL/min,250nm)indicated 93%ee:tR(major)=7.4min,tR(minor)=11.6min.
图10为本发明实施例8外消旋产物的高效液相色谱图,通过图10的外消旋产物的结构确定对照保留时间,然后得到实施例8制备产物的高效液相色谱图,如图11所示,ee值为93%。
实施例9
用1i代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2i,ee值为88%。
实施例9制备得到的产物2i的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
White solid.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.29–7.28(m,1H),7.12(d,J=7.0Hz,1H),7.04(t,J=7.5Hz,1H),6.75(t,J=7.5Hz,1H),6.67(d,J=7.5Hz,1H),6.26–6.25(m,1H),6.13(d,J=3.0Hz,1H),4.98(d,J=8.5Hz,1H),3.98(s,1H),3.76(q,J12=8.0Hz,J13=16.0Hz,1H),1.25(s,12H),1.23–1.08(m,1H),0.70–0.66(m,1H).13C NMR(125MHz,CDCl3)δ=155.31,150.08,141.64,133.32,127.24,123.80,118.92,109.98,108.89,107.06,83.18,61.81,42.14,24.93,24.87.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C19H25BNO3([M+H]+),326.1925,found 326.1958.[α]D19=-13.7,(c=1,CHCl3).HPLC analysis(AD-H,5%IPA/hexane,1mL/min,250nm)indicated 88%ee:tR(major)=7.9min,tR(minor)=15.6min
实施例10
用1j代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2j,ee值为83%。
实施例10制备得到的产物2j的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
Colorless oil.NMR Spectroscopy:1H NMR(400MHz,CDCl3)δ=7.34-7.27(m,4H),7.21(d,J=7.2Hz,2H),7.02(t,J=7.6Hz,1H),7.21(t,J=7.6Hz,2H),6.63(d,J=7.6Hz,1H),4.44(d,J=8.0Hz,1H),3.82(s,1H),3.62(q,J12=8.8Hz,J13=14.4Hz,1H),1.79(d,J=0.4Hz,3H),1.24(s,12H),1.10–1.03(m,1H),0.96–0.91(m,1H).13C NMR(100MHz,CDCl3)δ=150.24,137.86,136.94,134.43,130.89,128.98,128.01,127.31,126.64,126.18,124.05,118.40,108.68,83.16,71.21,40.47,24.95,16.23.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C24H31BNO2([M+H]+),376.2448,found 376.2442.[α]D19=+7.0,(c=0.5,CHCl3).HPLC analysis(AD-H,10%IPA/hexane,1mL/min,250nm)indicated 83%ee:tR(major)=5.6min,tR(minor)=8.9min.
实施例11
用1k代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2k。
图12为实施例11制备得到的2,3-二取代吲哚2k的1H-NMR的核磁共振谱;图13为实施例11制备得到的2,3-二取代吲哚2k的13C-NMR的核磁共振谱。谱图解析数据2k
White solid.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.09(t,J=3.5Hz,1H),7.00(s,1H),6.90(d,J=3.0Hz,2H),6.86(d,J=8.0Hz,1H),6.58(d,J=7.5Hz,1H),5.19(d,J=8.0Hz,1H),4.05(s,1H),3.64(q,J12=8.0Hz,J13=15.5Hz,1H),2.26(s,3H),1.24(d,J=3.0Hz,12H),1.06–1.01(m,1H),0.75–0.70(m,1H).13C NMR(125MHz,CDCl3)δ=147.30,144.61,133.76,128.38,127.58,126.19,125.04,125.01,123.85,109.14,83.12,64.45,42.77,25.01,24.78,20.93.Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C20H27BNO2S([M+H]+),356.1854,found 356.1862.[α]D27=+38.17,(c=1,CHCl3).HPLC analysis(AD-H,5%IPA/hexane,1mL/min,250nm)indicated 99%ee:tR(major)=8.9min,tR(minor)=9.9min.
图14为本发明实施例11外消旋产物的高效液相色谱图,通过图14的外消旋产物的结构确定对照保留时间,然后得到实施例11制备产物的高效液相色谱图,如图15所示,ee值为99%。
实施例12
用1l代替实例中的1a,其他条件同实例1,得到2,3-二取代吲哚类化合物2l,ee值为98%。
实施例12制备得到的产物2l的核磁氢谱数据、核磁碳谱数据和高效液相色谱数据如下:
Colorless oil.NMR Spectroscopy:1H NMR(500MHz,CDCl3)δ=7.11–7.10(m,1H),6.94–6.91(m,3H),6.75(dt,J=8.5Hz,1H),6.59–6.56(m,1H),5.22(s,1H),4.07(s,1H),3.66(q,J12=7.5Hz,J13=14.5Hz,1H),1.24(d,J=2.5Hz,12H),1.04–0.99(m,1H),0.76–0.72(m,1H).13C NMR(125MHz,CDCl3)δ=157.48(d,J=234.3Hz),144.66(d,J=188.63Hz),135.49,135.43,126.30,125.30,124.11,113.38,113.19,111.99,111.80,109.65,109.58,83.32,64.90,43.00,24.99,24.83.19FNMR(470MHz;CDCl3):δ=-127.50(s,1F).Mass Spectrometry:HRMS(ESI-TOF)(m/z):Calcd for C19H24BFNO2S([M+H]+),360.1603,found 360.1611.[α]D27=+39.62,(c=0.52,CHCl3).HPLC analysis(AD-H,Hexane/IPA=95/5,1mL/min,250nm)indicated 88%ee.After recrystallization,the ee value of the filtrate was98%:tR(major)=7.9min,tR(minor)=10.5min.
- 还没有人留言评论。精彩留言会获得点赞!