1,3-二氧代异吲哚啉苯甲酰胺类化合物及其用途的制作方法
本发明属于糖尿病治疗药物技术领域,具体涉及n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物、制备方法及其在制备治疗糖尿病药物中的应用。
背景技术:
糖尿病是当前严重威胁人类健康的重大疾病之一,正在临床应用的抗糖尿病药物多数以改善症状为主,无法彻底治愈该疾病。因此,亟需研发新一代的抗糖尿病药物。由于糖尿病主要是以胰岛β细胞数量减少和功能障碍为特征,因此靶向胰岛β细胞作为一种新颖的策略近年来受到广泛关注。该类研究对于新作用机制的抗糖尿病新药的研发具有极为重要的临床意义和应用前景。
在以胰岛β细胞为靶的多种策略中,抑制组蛋白去乙酰化酶3(hdac3)进而保护胰岛β细胞是一种具有前景的方向。值得一提的是,hdac3抑制剂brd3308已经在动物模型上表现出治疗糖尿病的良好效果。尽管如此,brd3308作为唯一的hdac3抑制剂类胰岛β细胞保护剂处于药物研究阶段,因而该化合物仍有可能存在成药性方面的不足而无法进入药物开发阶段。为此,有必要研究新结构的hdac3抑制剂类胰岛β细胞保护剂。
本专利涉及的n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物具有抑制hdac3的活性,且对棕榈酸诱导的小鼠胰岛nit-1细胞凋亡具有拮抗作用,有进一步研究的价值。截至目前,国内外均未报道过本专利涉及的化合物的保护胰岛β细胞的功能,因此该系列化合物属于新结构类型的药物苗头/先导结构。对该结构类型深入研究将有望发现治疗效果更为显著的新型糖尿病治疗药物。
技术实现要素:
本发明的目的是为了克服国内外市场上尚无hdac3抑制剂类胰岛β细胞保护剂作为糖尿病治疗药物的不足,提供一类抑制hdac3的n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物、制备方法及其其在制备治疗糖尿病药物中的应用。
本发明提供如下技术方案:
本发明第一方面提供了一种n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物及其药学上可接受的盐,它是如式(1)所示的化合物:
其中:
r选自氢、氟、氯、溴、甲基、乙基、丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基;
x选自氢、氟。
作为优选,本发明提供的n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物及其药学上可接受的盐为:
hbs-36:n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)-4-甲基苯甲酰胺;
hbs-58:n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)-4-乙基苯甲酰胺;
hbs-53:n-(2-氨基-4-氟苯基)-3-(1,3-二氧代异吲哚啉-2-基)-苯甲酰胺;
hbs-55:n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)-4-氟苯甲酰胺。
本发明的第二方面提供了n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物的制备方法,它包括如下步骤:
a:
其中,x选自氢,r选自氢、氟、氯、溴、甲基、乙基、丙基、异丙基、正丁基、仲丁基、异丁
基、叔丁基;
i.中间体imt1的制备:1)将邻苯二甲酸酐和取代3-氨基苯甲酸溶于一定体积的乙酸中,150℃微波反应30分钟,反应液中加少许水,析出沉淀,过滤并干燥,得中间体imt1;或者2)将邻苯二甲酸和取代3-氨基苯甲酸溶于一定体积的乙腈中,150℃微波反应30分钟,反应液中加少许乙腈,析出沉淀,过滤并干燥,得中间体imt1;
ii.目标化合物的制备方法:将中间体imt1与邻苯二甲酰丁辛酯溶于n,n-二甲基甲酰胺,在氮气保护下缓慢滴加三乙胺,室温搅拌15分钟,滴加邻苯二胺的n,n-二甲基甲酰胺溶液反应,薄层色谱法监测反应结束后,将反应液倒入乙酸乙酯中,并加入蒸馏水,静置分层。有机层经饱和氯化钠溶液洗涤、无水硫酸镁干燥后过滤,滤液减压浓缩,残余物经60-100目拌样,硅胶柱层析纯化,得到目标化合物;
或者b:
其中,x选自氟,r选自氢、氟、氯、溴、甲基、乙基、丙基、异丙基、正丁基、仲丁基、异丁
基、叔丁基;
i.中间体imt1的制备方法同步骤a;
ii.中间体imt2的制备:
①5-氟-2-硝基苯胺的二氯甲烷溶液、二碳酸二叔丁酯及4-二甲氨基吡啶室温下搅拌反应12小时后,将反应液倒入二氯甲烷中,加入蒸馏水,静置分层,分出二氯甲烷层,二氯甲烷层经饱和氯化钠溶液洗涤、无水硫酸钠干燥并过滤后,滤液减压浓缩得产物;加入二氯甲烷溶解产物,与三氟乙酸反应,薄层色谱法监测反应结束后,反应液中加入饱和碳酸氢钠溶液,室温搅拌15分钟,加入蒸馏水,有机层经饱和氯化钠溶液洗涤、无水硫酸钠干燥后过滤,滤液减压浓缩,进一步使用正己烷重结晶,得5-氟-2-硝基苯基氨基甲酸叔丁酯纯品;
②将5-氟-2-硝基苯基氨基甲酸叔丁酯溶于四氢呋喃,加入钯碳催化剂后加热回流,加入甲酸铵水溶液反应,薄层色谱法监测反应完全后,过滤,减压蒸去四氢呋喃,水相经二氯甲烷萃取,有机层经饱和氯化钠溶液洗涤、无水硫酸钠干燥后过滤、滤液减压浓缩,得中间体2-氨基-5-氟苯基氨基甲酸叔丁酯纯品imt2;
iii.目标化合物的制备方法:将中间体imt1与邻苯二甲酰丁辛酯溶于n,n-二甲基甲酰胺,在氮气保护下缓慢滴加三乙胺,室温搅拌15分钟,滴加imt2的n,n-二甲基甲酰胺溶液反应,薄层色谱法监测反应结束后,将反应液倒入乙酸乙酯中,加入蒸馏水,静置分层,有机层经饱和氯化钠溶液洗涤、无水硫酸镁干燥后过滤,滤液减压浓缩;将所得固体溶于二氧六环溶液中,并滴加4mol/l的盐酸/二氧六环溶液,反应3小时后,将反应液倒入至乙酸乙酯中,加入蒸馏水,静置分层;有机层经饱和氯化钠洗涤、无水硫酸镁干燥后过滤,滤液减压浓缩,残余物经硅胶柱层析纯化,得到目标化合物;
acoh为乙酸,mecn为乙腈,microwave为微波,bop为邻苯二甲酰丁辛酯,dmf为n,n-二甲基甲酰胺,net3为三乙胺,dmap为4-二甲氨基吡啶,(boc)2o为二碳酸二叔丁酯,tfa为三氟乙酸,hcoonh4为甲酸铵,pd/c为钯碳催化剂,hcl/dioxane为盐酸/二氧六环溶液。
本发明第三方面提供了n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物及其药学上可接受的盐在制备治疗糖尿病药物中的应用。
作为优选,治疗糖尿病药物是通过保护胰岛β细胞产生治疗效果的。
作为优选,保护胰岛β细胞为通过抑制组蛋白去乙酰化酶3而产生的治疗效果。
优选的,治疗效果包括对棕榈酸诱导的小鼠胰岛nit-1细胞凋亡的拮抗作用。
本发明通过体外细胞实验证实,若干n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物可拮抗棕榈酸诱导的小鼠胰岛nit-1细胞凋亡,显著提高胰岛β细胞活力。
本发明第四方面提供了一种药物组合物,其特征在于,所述的药物组合物包括本发明第一方面所述的n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物及其药学上可接受的盐以及药学上可接受的载体或赋形剂。所述的药物组合物选自注射剂、片剂、丸剂、胶囊、悬浮剂或乳剂。所述载体包括药学领域常规的赋形剂、粘合剂、湿润剂、崩解剂、吸收促进剂、表面活性剂、吸附载体、润滑剂等。
作为优选,该药物给药剂型为注射剂、片剂、丸剂、胶囊、悬浮剂或乳剂,给药途径为口服、经皮、静脉或肌肉注射。
与现有技术相比,本发明具有如下有益效果:
体外生物活性测试表明,n-(2-氨基苯基)-3-(1,3-二氧代异吲哚啉-2-基)苯甲酰胺类化合物可以改善胰岛β细胞功能,为首次公开。该系列化合物有进一步研发成为新型胰岛β细胞类抗糖尿病药物的潜力。
附图说明
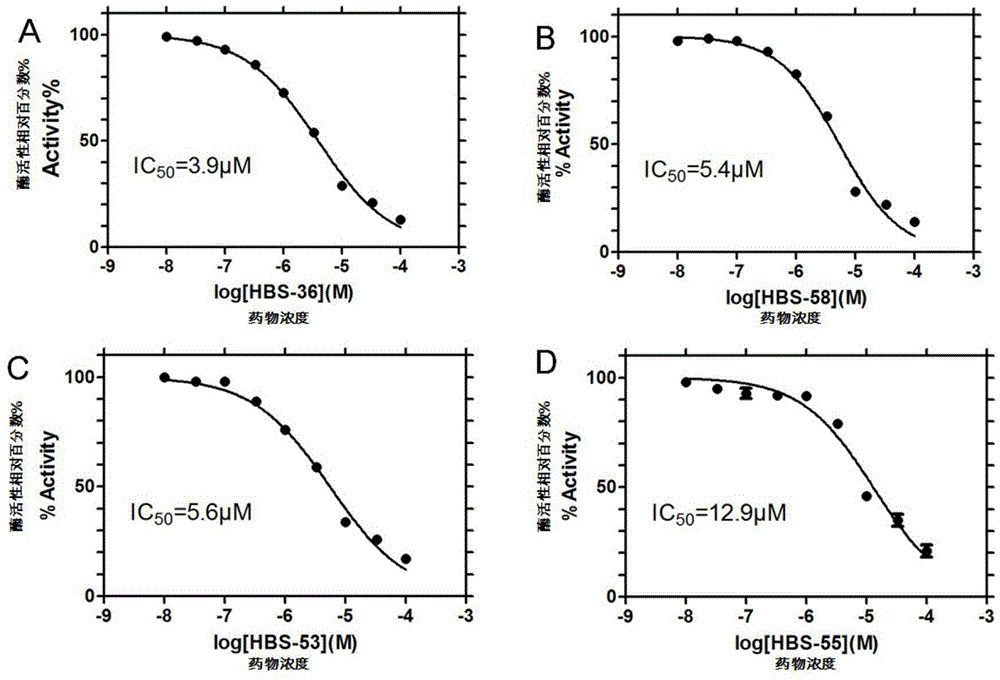
图1化合物抑制hdac3的量效曲线及ic50
具体实施方式
下面结合具体实施例对本发明作进一步阐述,但实施例仅用于说明本发明,而不是对本发明进行限制。实施例中所用实验方法如无特殊说明,均为常规方法;所使用的材料、试剂等如无特殊说明,为可从商业途径得到的试剂和材料。
一、化合物的制备和检测
实施例1:化合物hbs-36的制备和检测
往10-20ml微波反应管中依次加入498.39mg(3mmol)邻苯二甲酸与453.48mg(3mmol)3-氨基-4-甲基苯甲酸及3.5ml乙腈,180℃微波条件下反应30分钟。随后取出微波反应管,并往管中加入少许乙腈,过滤,滤饼于真空干燥箱(50℃)中干燥6小时,得中间体3-(1,3-二氧代异吲哚啉-2-基)-4-甲基苯甲酸。
取247.34mg(0.88mmol)的3-(1,3-二氧代异吲哚啉-2-基)-4-甲基苯甲酸与468mg(1.06mmol)的邻苯二甲酰丁辛酯溶于n,n-二甲基甲酰胺中,在氮气保护下滴加490μl(3.52mmol)三乙胺,室温搅拌15分钟,然后滴加114.2mg(1.06mmol)邻苯二胺的n,n-二甲基甲酰胺溶液,反应2小时,薄层色谱法监测原料消失且有新物质产生,lc-ms监测其为产物峰。停止反应,将反应液倒入20ml乙酸乙酯中,并分两次加入100ml蒸馏水,静置分层。有机层经100ml饱和氯化钠溶液洗涤2次,无水硫酸镁干燥,过滤,滤液减压浓缩,残余物经60-100目拌样,硅胶柱层析(洗脱剂:pe-acoet)纯化,得到164mg固体样品(hbs-36,收率为50.2%)。
终产物结构表征如下:1hnmr(500mhz,dmso-d6)δ9.74(s,1h),8.10(d,j=8.0hz,1h),8.01-8.06(m,3h),7.95-7.99(m,2h),7.59(d,j=8.0hz,1h),7.19(d,j=7.8hz,1h),7.00(t,j=7.6hz,1h),6.81(d,j=8.1hz,1h),6.62(t,j=7.5hz,1h),4.95(s,2h),2.25(s,3h);13cnmr(126mhz,dmso-d6)δ167.62,164.92,143.94,140.76,135.52,133.99,132.30,131.69,131.27,129.68,128.97,127.46,127.23,124.29,123.72,116.82,116.72,18.18;hrmscalcdforc22h18o3n3[m+h]+372.13427,found:372.13336.
实施例2:化合物hbs-58的制备和检测
往10-20ml微波反应管中依次加入444.36mg(3mmol)邻苯二甲酸酐与495.57mg(3mmol)3-氨基-4-乙基苯甲酸及3.5ml乙酸,150℃微波条件下反应30分钟。随后取出微波反应管,并往管中加入少许水,过滤,滤饼于真空干燥箱(50℃)中干燥6小时,得中间体3-(1,3-二氧代异吲哚啉-2-基)-4-乙基苯甲酸。
取295.08mg(1mmol)的3-(1,3-二氧代异吲哚啉-2-基)-4-乙基苯甲酸与530.74mg(1.2mmol)邻苯二甲酰丁辛酯溶于n,n-二甲基甲酰胺,在氮气保护下滴加560μl(4mmol)三乙胺,室温搅拌15分钟,然后滴加129.8mg(1.2mmol)邻苯二胺的n,n-二甲基甲酰胺溶液,反应2小时,薄层色谱法监测原料消失且有新物质产生,lc-ms监测其为产物峰。停止反应,将反应液到入20ml乙酸乙酯中,并分两次加入100ml蒸馏水,静置分层。有机层经100ml饱和氯化钠溶液洗涤2次,无水硫酸镁干燥,过滤,滤液减压浓缩,残余物经60-100目拌样,硅胶柱层析(洗脱剂:pe-acoet)纯化,得到222mg固体(hbs-58,收率为57.6%)。
终产物结构表征如下:1hnmr(500mhz,dmso-d6)δ9.81(s,1h),8.15(d,j=8.1hz,1h),8.00-8.08(m,3h),7.93-8.00(m,2h),7.63(d,j=8.1hz,1h),7.23(d,j=7.8hz,1h),7.04(t,j=7.6hz,1h),6.87(d,j=8.0hz,1h),6.71(t,j=7.5hz,1h),2.57(q,j=7.6hz,2h),1.14(t,j=7.6hz,3h);13cnmr(101mhz,dmso-d6)δ167.83,164.81,146.42,142.21,135.38,133.79,132.06,130.92,129.84,129.54,129.17,127.28,127.08,124.32,124.11,117.78,117.28,24.37,14.82;hrmscalcdforc23h20o3n3[m+h]+386.14992,found:386.14880。
实施例3:化合物hbs-55的制备和检测
往10-20ml微波反应管中依次加入444.36mg(3mmol)邻苯二甲酸酐与465.39mg(3mmol)的3-氨基-4-氟苯甲酸及3.5ml乙酸,150℃微波条件下反应30分钟。随后取出微波反应管,并往管中加入少许水,过滤,滤饼于真空干燥箱(50℃)中干燥6小时,得中间体3-(1,3-二氧代异吲哚啉-2-基)-4-氟苯甲酸。
取301.01mg(1mmol)3-(1,3-二氧代异吲哚啉-2-基)-4-氟苯甲酸与530.74mg(1.2mmol)的邻苯二甲酰丁辛酯溶于n,n-二甲基甲酰胺,在氮气保护下缓慢滴加560μl(4mmol)三乙胺,室温搅拌15分钟,然后滴加含129.8mg(1.2mmol)邻苯二胺的n,n-二甲基甲酰胺溶液,反应2小时,薄层色谱法监测原料消失且有新物质产生,lc-ms监测其为产物峰。停止反应,将反应液到入20ml乙酸乙酯中,并分两次加入100ml蒸馏水,静置分层。有机层经100ml饱和氯化钠溶液洗涤2次,无水硫酸镁干燥,过滤,滤液减压浓缩,残余物经60-100目拌样,硅胶柱层析(洗脱剂:pe-acoet)纯化,得到150mg固体(hbs-55,收率为40.0%)。
终产物结构表征如下:1hnmr(500mhz,dmso-d6)δ9.84(s,1h),8.25-8.35(m,2h),8.06(d,j=4.1hz,2h),7.99(d,j=4.2hz,2h),7.67(t,j=9.2hz,1h),7.21(d,j=7.8hz,1h),7.03(t,j=7.7hz,1h),6.84(d,j=8.0hz,1h),6.66(t,j=7.6hz,1h),5.28(s,2h);13cnmr(101mhz,dmso-d6)δ166.67,164.07,135.59,132.21,132.18,131.92,131.44,131.13,131.04,127.41,127.23,124.31,123.62,119.74,117.10,116.78,116.66;hrmscalcdforc21h15o3n3f[m+h]+376.10920,found:376.10834。
实施例4:化合物hbs-53的制备和检测
往150ml双颈瓶中依次加入60ml含9.4g(60mmol)5-氟-2-硝基-苯胺的二氯甲烷溶液,26.6g(121.2mmol)二碳酸二叔丁酯及2.6g(20mmol)4-二甲氨基吡啶,室温下搅拌12小时后停止反应。将反应液倒入60ml二氯甲烷中,并分两次加入100ml蒸馏水,静置分层。有机层经100ml饱和氯化钠溶液洗涤2次,无水硫酸钠干燥,过滤,滤液减压浓缩得产物。加入50ml二氯甲烷以溶解产物,并加入7.9ml(105.6mmol)三氟乙酸,利用薄层色谱法监测反应,约6小时后停止反应。反应液中加入100ml饱和碳酸氢钠溶液,室温搅拌15分钟,再加入蒸馏水萃取2次,静置分层,取有机层。有机层经饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,经60ml正己烷重结晶,得9g5-氟-2-硝基苯基氨基甲酸叔丁酯纯品。
往100ml双颈瓶中依次加入40ml四氢呋喃、7.7g(30mmol)5-氟-2-硝基苯基氨基甲酸叔丁酯及5g钯碳,加热回流,然后加入60ml含19g(300mmol)甲酸铵的水溶液,利用薄层色谱法监测反应进程。1小时后显示原料消失,过滤,减压蒸去四氢呋喃,水相经60ml二氯甲烷萃取,有机层再用饱和氯化钠溶液(100ml×2)洗涤,并经无水硫酸钠干燥,过滤,滤液减压浓缩,得5.89g中间体2-氨基-5-氟苯基氨基甲酸叔丁酯,收率为86.9%。esi-ms(m/z):227.24[m+h]+。
往10-20ml微波反应管中依次加入498.39mg(3mmol)的邻苯二甲酸与411.42mg(3mmol)的3-氨基苯甲酸及3.5ml乙腈,180℃微波条件下反应30分钟。随后取出微波反应管,并往管中加入少许乙腈,过滤,滤饼于真空干燥箱(50℃)中干燥6小时,得中间体3-(1,3-二氧代异吲哚啉-2-基)苯甲酸。
取267.05mg(1mmol)3-(1,3-二氧代异吲哚啉-2-基)苯甲酸与530.74mg(1.2mmol)邻苯二甲酰丁辛酯溶于n,n-二甲基甲酰胺,在氮气保护下滴加560μl(4mmol)三乙胺,室温搅拌15分钟,然后滴加含271.34mg(1.2mmol)2-氨基-5-氟苯基氨基甲酸叔丁酯的n,n-二甲基甲酰胺溶液,反应2小时,薄层色谱法监测原料消失且有新物质产生,lc-ms监测其为产物峰。停止反应,将反应液到入20ml乙酸乙酯中,并加入100ml蒸馏萃取2次,有机层再经饱和氯化钠溶液(50ml×2)洗涤,无水硫酸镁干燥,过滤,滤液减压浓缩。将所得固体溶于二氧六环溶液中,并滴加4ml4mol/l的hcl/二氧六环溶液,反应3小时后,薄层色谱法监测原料消失,而且有新物质产生,lc-ms监测到产物峰。停止反应,将反应液到入20ml乙酸乙酯中,并加入100ml蒸馏水水洗2次,静置分层,取有机层。有机层经100ml饱和氯化钠溶液分2次洗涤(50ml×2),无水硫酸镁干燥,过滤,滤液减压浓缩,残余物经硅胶柱层析(洗脱剂:pe-acoet)纯化,得到160mg固体(hbs-53,收率为43.0%)。
终产物结构表征如下:1hnmr(500mhz,dmso-d6)δ9.72(s,1h),8.12(d,j=6.4hz,1h),8.08(s,1h),7.99-8.06(m,2h),7.92-7.99(m,2h),7.64-7.74(m,2h),7.16(t,j=7.6hz,1h),6.58(dd,j=11.2,2.9hz,1h),6.40(td,j=8.4,2.8hz,1h),5.31(s,2h);13cnmr(101mhz,dmso-d6)δ167.42,165.24,160.40,146.10,135.84,135.27,132.52,132.03,130.87,129.30,127.74,127.56,123.98,102.57,101.73;hrmscalcdforc21h15o3n3f[m+h]+376.10920,found:376.10828。
三、化合物的生物活性评价
实验例1:hdac3酶抑制活性测定
1.材料:全长的重组人源gst标记的hdac3/ncor2酶(残基237-489)购自加拿大英属哥伦比亚的signalchem公司;hdac3/ncor2酶的底物为10μm的ac-leu-glylys(ac)-amc,由上海glbiochem公司合成;胰酶购自上海sangonbiotech公司。活性测试使用的缓冲液的组成包括:25mmtris、ph8.0、1mmmgcl2、0.1mg/mlbsa、137mmnacl和2.7mmkcl。待测化合物先用dmso溶解制备母液(10mm),再使用缓冲液稀释到10倍于待测浓度值的浓度。
2.方法:首先,测定10μm浓度的待测化合物对hdac3亚型的抑制率;进一步确定抑制率高于50%的化合物的量效曲线与ic50值。具体操作如下:酶学反应在nunc的96孔板进行。往每一个孔中加入25μl的缓冲液(含荧光底物)、10μl待测化合物的溶液和15μl的hdac3/ncor2酶,然后将96孔板在37℃下孵育30分钟。接着,往每个孔内加入50μl0.4mg/ml的显影剂胰酶,然后再次将孔板于37℃下孵育30分钟。本实验使用hdac3抑制剂saha作为阳性对照,不加待测化合物作为阴性对照。使用perkin-elmerenspire酶标仪检测荧光信号,激发波长为360nm,发射波长为460nm。将荧光值代入如下公式,计算活性百分率:%酶活性={(fl待测化合物–fl本底)/(fl酶–fl本底)}×100%,然后用graphpadprism5软件处理,以归一化的量效拟合方式的非线性回归拟合量效关系曲线,并计算出待测化合物的ic50值。
3.10μm浓度的代表性化合物对hdac3的抑制率见表1。其中,阳性对照saha的ic50=46.1nm。
4.代表性化合物抑制hdac3的量效曲线和ic50见图1。结果显示,hbs-36的ic50值为3.9μm(图1a),hbs-58的ic50值为5.4μm(图1b),hbs-53的ic50值为5.6μm(图1c),hbs-55的ic50值为12.9μm(图1d)。
5.各化合物量效曲线对应的原始数据见表2至表5。
实验例2:化合物对棕榈酸(palmiticacid,pa)诱导的小鼠胰岛nit-1细胞凋亡的拮抗作用
1.细胞培养
nit-1细胞,atcc产品,购自于中国医学科学院基础医学研究所细胞库。细胞复苏后接种于t25细胞瓶中,培养基为dmem/f12,37℃、5%co2培养箱中培养,待细胞生长至汇合度为80%左右时传代继续培养用于后续细胞实验。
待细胞生长至汇合度为80%左右时,消化细胞用吹打管至单细胞混悬液,细胞计数,稀释成3×105个/ml,接种于96孔板中,3×104个/100μl/well,培养24h后用于实验。
2.细胞活力检测
nit-1细胞诱导凋亡的条件为0.5mmpa,诱导时间为12h。在诱导凋亡的过程中,检测细胞活力反映细胞凋亡程度。nit-1细胞在96孔板中培养24h后,更换培养基(含1%牛血清白蛋白的dmem培养基,25mm葡萄糖),设置培养基对照组(pa-)。培养基中含0.5mmpa组(pa+),并加入不同的化合物(配制化合物相应的浓度)孵育,加入溶媒dmso为阴性对照组。以上条件培养细胞12h后,每孔加入5μl的cck8细胞活性检测试剂,于450nm处测定od值,统计各组差异,p<0.05具有统计学差异。
3.化合物拮抗棕榈酸诱导的小鼠胰岛nit-1细胞凋亡的结果见表6。
表1:代表性化合物在浓度为10μm时对hdac3的酶抑制率。
表2:不同浓度的hbs-36对应的hdac3酶活性(发光强度)相对百分数
表3:不同浓度的hbs-58对应的hdac3酶活性(发光强度)相对百分数
表4:不同浓度的hbs-53对应的hdac3酶活性(发光强度)相对百分数
表5:不同浓度的hbs-55对应的hdac3酶活性(发光强度)相对百分数
表6:代表性化合物在浓度为10μm时拮抗棕榈酸诱导的小鼠胰岛nit-1细胞凋亡的作用
a:od值越大代表细胞活力越强
*p<0.05,**p<0.01,***p<0.001vspa+;***(b)p<0.001vspa-。
- 还没有人留言评论。精彩留言会获得点赞!