水溶性β-环糊精酰胺化衍生物及合成方法和在抗氧化、抗菌方面的应用与流程
本发明属于有机合成与精细化工技术领域,具体涉及一种水溶性β-环糊精酰胺化衍生物,以及该化合物的合成方法和在抗氧化、抗菌方面的应用。
背景技术
环糊精(cyclodextrin,cd)主要是指环糊精糖基转移酶作用于淀粉得到的降解产物。环糊精是典型的可再生、无毒、可生物降解资源,并且具有“内腔疏水,外壁亲水”的特殊结构和性质,已成为构筑各种功能材料的优良结构单元,其中β-环糊精价廉易得,应用最广。
环糊精在水中的溶解度极小,对药物客体分子的识别受到了阻碍,限制了其在药物学领域的应用,因而β-环糊精衍生物的合成越来越受到人们的青睐。β-环糊精衍生物在水中的溶解性能会显著地增强,这样也能增加空腔的尺寸,使其与客体药物的包络作用增强。
国内外对环糊精进行改性主要有两种途径:化学法和酶工程法。化学法是使环糊精分子表面的醇羟基发生各种化学反应,如酯化、氧化、醚化、交联等化学反应,从而使环糊精的分子表面与新的官能团相连,获得具有不同性质的环糊精衍生物,是最主要的改性方法。对天然环糊精进行改性后得到的环糊精衍生物具备与母体环糊精不同的特性,特别是环糊精的6位羟基被取代所得到的衍生物和客体分子之间的结合能力更强。酶工程法是制备支链环糊精的方法,即在环糊精葡萄糖基转移酶或普鲁蓝酶的作用下将单糖或低聚糖结合到环糊精上构成所需的支链环糊精。环糊精衍生物的成本低、相溶性好,扩大了应用范围、发掘了新的用途,能很好地应用于制药、分析、分离、催化等各个方面。因此,有必要对环糊精衍生物进行研究。
技术实现要素:
本发明所要解决的技术问题在于提供一种同时具备抗氧化、抗菌性能的水溶性β-环糊精酰胺化衍生物,以及该化合物的合成方法和应用。
解决上述技术问题所采用的水溶性β-环糊精酰胺化衍生物的结构式如下所示:
式中r1、r2、r3、r4、r5各自独立地代表h、oh、c1~c2烷氧基、c1~c3烷基中任意一种,且其中至少有一个为oh;
本发明水溶性β-环糊精酰胺化衍生物合成路线和具体合成方法如下所示:
1、合成β-环糊精磺酰化衍生物
以氢氧化钠水溶液为溶剂,将β-环糊精与对甲苯磺酰氯按摩尔比为0.6:1,室温反应3小时后抽滤,滤液用盐酸将ph调节至2,分离纯化产物,得到式i所示的β-环糊精磺酰化衍生物。
2、合成β-环糊精氨基化衍生物
以无水乙醇为溶剂,将β-环糊精磺酰化衍生物与乙二胺按摩尔比为1:290~300,80℃下反应4小时,分离纯化产物,得到式ii所示的β-环糊精氨基化衍生物。
3、合成水溶性β-环糊精酰胺化衍生物
以n,n-二甲基甲酰胺为溶剂,使用1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和1-羟基苯并三唑作为缩合剂活化式iii所示的肉桂酸衍生物,再加入β-环糊精氨基化衍生物,室温反应,反应完后分离纯化产物,得到β-环糊精酰胺化衍生物。
上述步骤3中,所述肉桂酸衍生物与1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、1-羟基苯并三唑、β-环糊精氨基化衍生物的摩尔比为1:0.9~1:0.9~1:0.6~1。
本发明水溶性β-环糊精酰胺化衍生物作为抗氧化剂在抗氧化方面的应用。
本发明水溶性β-环糊精酰胺化衍生物作为抗菌剂在抗菌方面的应用。
与现有技术相比,本发明具有以下有益效果:
1、本发明水溶性β-环糊精酰胺化衍生物的合成方法原料廉价易得、操作简单且条件温和,所合成的β-环糊精酰胺化衍生物纯度较高。
2、本发明β-环糊精酰胺化衍生物水溶性十分良好,大于600mg/ml,而β-环糊精母体水溶性仅为18.5mg/ml。
3、本发明采用肉桂酸衍生物改性β-环糊精在改善环糊精母体水溶性的同时,可以实现多功能化,仅凭β-环糊精酰胺化衍生物主体就具备良好的抗氧化、抗菌性能。而一般的环糊精衍生物主体需要包合功能性客体分子才能具备附加功能。本发明β-环糊精酰胺化衍生物扩大了应用范围、发掘了新的用途,可用于医学、日用化工、食品工业等领域。
附图说明
图1是实施例1制备的β-环糊精磺酰化衍生物的质谱图。
图2是实施例1制备的β-环糊精磺酰化衍生物的核磁共振氢谱图。
图3是实施例1制备的β-环糊精氨基化衍生物的质谱图。
图4是实施例1制备的β-环糊精氨基化衍生物的核磁共振氢谱图。
图5是实施例1制备的β-环糊精酰胺化衍生物的质谱图。
图6是实施例1制备的β-环糊精酰胺化衍生物的核磁共振氢谱图。
图7是实施例2制备的β-环糊精酰胺化衍生物的质谱图。
图8是实施例2制备的β-环糊精酰胺化衍生物的核磁共振氢谱图。
图9是实施例3制备的β-环糊精酰胺化衍生物的质谱图。
图10是实施例3制备的β-环糊精酰胺化衍生物的核磁共振氢谱图。
图11是实施例1制备的不同浓度β-环糊精酰胺化衍生物添加相同浓度dpph的吸光度曲线。
图12是实施例2制备的不同浓度β-环糊精酰胺化衍生物添加相同浓度dpph的吸光度曲线。
图13是实施例3制备的不同浓度β-环糊精酰胺化衍生物添加相同浓度dpph的吸光度曲线。
图14是实施例1~3制备的β-环糊精酰胺化衍生物分别添加相同浓度dpph的吸光度曲线。
图15是实施例1~3制备的β-环糊精酰胺化衍生物对金黄色葡萄球菌的杀菌率图表。
具体实施方式
下面结合附图和实施例对本发明进一步详细说明,但本发明的保护范围不仅限于这些实施例。
实施例1
合成结构式如下的β-环糊精酰胺化衍生物
1、合成β-环糊精磺酰化衍生物
将17.22g(0.015mol)β-环糊精溶于200ml0.25mol/l氢氧化钠水溶液中,并向溶液中滴加16.5ml溶有4.35g(0.023mol)对甲苯磺酰氯的乙腈溶液,在室温下反应3小时,抽滤,滤液用盐酸将ph调至2后,在4℃条件下静置过夜,有大量白色固体析出,抽滤,用去离子水重结晶,40℃条件下真空干燥5小时,得到白色粉末状产品β-cd-6-ots。
所得β-cd-6-ots采用质谱仪和核磁共振谱仪对结构进行表征,结果见图1和图2。该化合物的相对分子量为1310.3606,由图1可见,其质谱分子离子峰为1311.3664,对应的离子峰为m+;图2中,化合物中的每种氢均可与氢谱上的化学位移和积分一一对应,具体数据为:1hnmr(400mhz,dmso):δ:2.41(s,3h,ch3),3.24-3.80(m,42h,ch),4.47-4.55(m,6h,oh),4.80-4.84(m,7h,ch),5.62-5.83(m,14h,oh),7.42(d,2h,phh),7.74(d,2h,phh).
2、合成β-环糊精氨基化衍生物
取3g(0.0023mol)β-cd-6-ots加入溶有45ml(0.675mol)乙二胺的乙醇溶液中,搅拌溶解后升温至80℃,反应4小时,减压蒸馏除溶剂。将所得固体使用少量热水溶解,加入300ml丙酮洗涤,抽滤。重复提纯3次,40℃条件下真空干燥5小时,得到白色粉末状产品β-cd-6-eda。
所得β-cd-6-eda采用质谱仪和核磁共振谱仪对结构进行表征,结果见图3和图4。该化合物的相对分子量为1176.4279,由图3可见,其质谱分子离子峰为1177.4312,对应的离子峰为m+;图4中,化合物中的每种氢均可与氢谱上的化学位移和积分一一对应,具体数据为:1hnmr(400mhz,dmso):δ:2.09-2.01(m,1h,nh),2.32-2.34(m,2h,ch2),2.43-2.45(m,2h,ch2),3.24-3.80(m,42h,ch),4.47-4.55(m,6h,oh),4.80-4.84(m,7h,ch),5.62-5.83(m,14h,oh),8.20(t,2h,nh2).
3、合成β-环糊精酰胺化衍生物
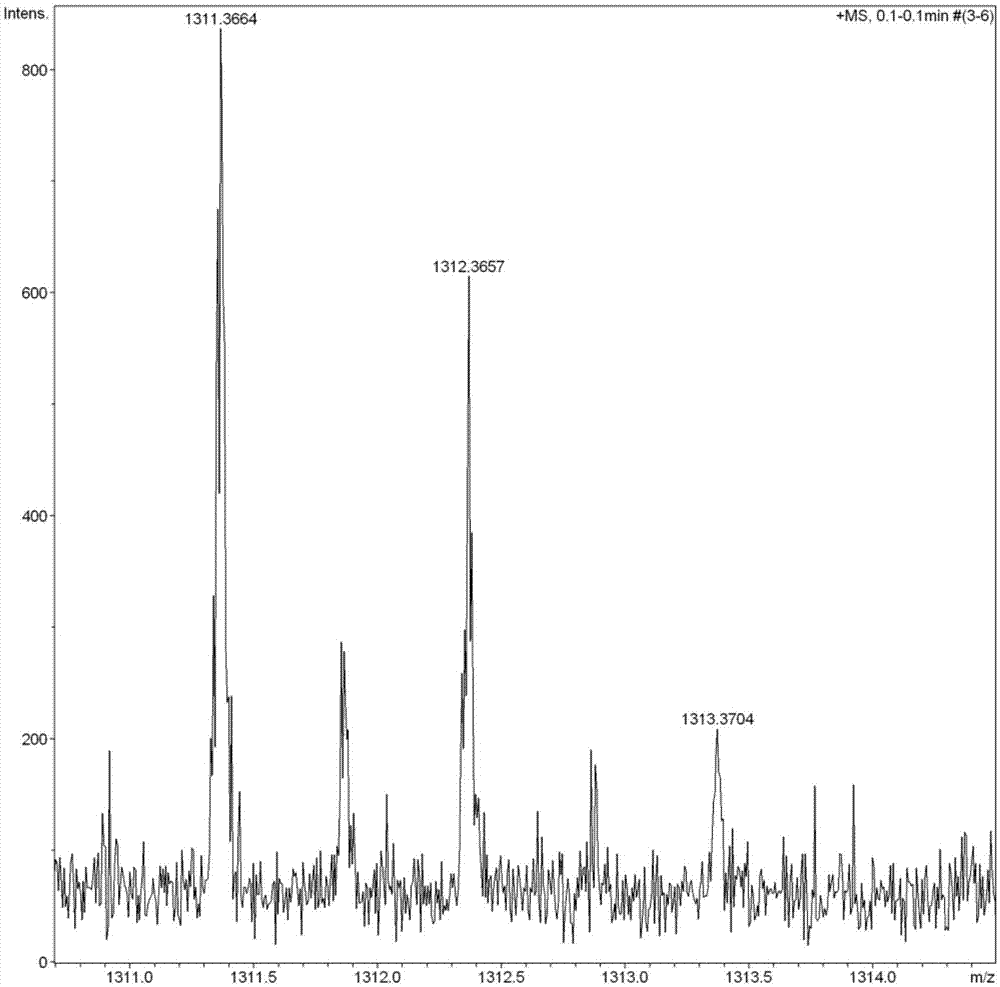
取0.29g(0.15mmol)阿魏酸(fa)、0.25g(0.13mmol)1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、0.176g(0.13mmol)1-羟基苯并三唑加入溶有865μl(0.68mmol)三乙胺的n,n-二甲基甲酰胺中,在冰浴条件下反应2小时。反应完后,缓慢加入10ml溶解有1.35g(0.10mmol)β-cd-6-eda的无水n,n-二甲基甲酰胺溶液,室温反应5天,用丙酮洗涤,柱层析分离(洗脱液为正丙醇:乙醇:水:氨水=12:2:4:1的混合液),得到β-环糊精酰胺化衍生物,记为β-cd-6-fa,其产率为56.58%。
所得β-cd-6-fa采用质谱仪和核磁共振谱仪对结构进行表征,结果见图5和图6。该化合物的相对分子量为1352.4753,由图5可见,其质谱分子离子峰为1353.4829,对应的离子峰为m+;图6中,化合物中的每种氢均可与氢谱上的化学位移和积分一一对应,具体数据为:1hnmr(400mhz,dmso):δ:2.09-2.01(m,1h,nh),2.32-2.34(m,2h,ch2),2.43-2.45(m,2h,ch2),3.24-3.72(m,42h,ch),3.80(s,3h,ch3),4.47-4.55(m,6h,oh),4.80-4.84(m,7h,ch),5.62-5.83(m,14h,oh),6.46(d,1h,ch),6.81(d,1h,phh),7.00(d,1h,phh),7.13(s,1h,phh),7.31(d,1h,ch),7.93(s,1h,oh),8.20(t,h,nh).
实施例2
合成结构式如下的β-环糊精酰胺化衍生物
本实施例中,用等摩尔香豆酸(p-ca)替换实施例1中的阿魏酸,其他步骤与实施例1相同,得到的黄色固体β-环糊精酰胺化衍生物,记为β-cd-6-p-ca,其产率为50.71%。
所得β-cd-6-p-ca采用质谱仪和核磁共振谱仪对结构进行表征,结果见图7和图8。该化合物的相对分子量为1322.4720,由图7可见,其质谱分子离子峰为1323.4710,对应的离子峰为m+;图8中,化合物中的每种氢均可与氢谱上的化学位移和积分一一对应,具体数据为:1hnmr(400mhz,dmso):δ:2.09-2.01(m,1h,nh),2.32-2.34(m,2h,ch2),2.43-2.45(m,2h,ch2),3.24-3.80(m,42h,ch),4.47-4.55(m,6h,oh),4.80-4.84(m,7h,ch),5.62-5.83(m,14h,oh),6.44(d,1h,ch),6.78(d,2h,phh),7.30(d,1h,ch),7.38(d,2h,phh),7.93(s,1h,oh),8.18(t,1h,nh).
实施例3
合成结构式如下β-环糊精酰胺化衍生物
本实施例中,用等摩尔咖啡酸(ca)替换实施例1中的阿魏酸,其他步骤与实施例1相同,得到的黄色固体β-环糊精酰胺化衍生物,记为β-cd-6-ca,其产率为60.12%。
所得β-cd-6-ca采用质谱仪和核磁共振谱仪对结构进行表征,结果见图9和图10。该化合物的相对分子量为1338.4669,由图9可见,其质谱分子离子峰为1339.4659,对应的离子峰为m+;图10中,化合物中的每种氢均可与氢谱上的化学位移和积分一一对应,具体数据为:1hnmr(400mhz,dmso):δ:2.09-2.01(m,1h,nh),2.32-2.34(m,2h,ch2),2.43-2.45(m,2h,ch2),3.24-3.80(m,42h,ch),4.47-4.55(m,6h,oh),4.80-4.84(m,7h,ch),5.62-5.83(m,14h,oh),6.30-6.35(d,1h,ch),6.72-6.74(d,1h,phh),6.82-6.86(d,1h,phh),6.94(s,1h,phh),7.20-7.23(d,1h,ch),7.94(s,1h,oh),7.98(s,1h,oh),8.20(t,1h,nh).
实施例4
实施例1~3制备的β-环糊精酰胺化衍生物作为抗氧化剂的应用
取7.20mgdpph用乙醇定容于100ml容量瓶中,超声溶解后,分别移取5mldpph溶解于比色管中,然后分别加入0、0.2、0.4、0.6、0.8、1ml1mmol/lβ-环糊精酰胺化衍生物水溶液,并用乙醇和水体积比为1:1的混合液定容至6ml,体系中β-环糊精酰胺化衍生物的浓度按加入的体积由少到多依次为0、33、66、99、132、165μmol/l。避光静置30分钟,测试uv-vis,结果见图11~14。
从图11~13中可以看出,随着β-环糊精酰胺化衍生物浓度的升高,dpph在517nm处的吸光度逐渐降低,并且517nm吸光度的变化呈现较好的线性关系。由图14可见,33μmol/lβ-cd-6-fa对dpph自由基清除率为26.38%,33μmol/lβ-cd-6-p-ca对dpph自由基清除率为8.96%,33μmol/lβ-cd-6-ca对dpph自由基清除率为99.91%,说明在相同浓度下,β-cd-6-ca的抗氧化性能最好。而当β-cd-6-fa浓度为198μmol/l、β-cd-6-p-ca浓度为330μmol/l时,对dpph自由基最高清除率也可达到99.00%以上。
实施例5
实施例1~3制备的β-环糊精酰胺化衍生物作为抗菌剂的应用
取1ml金黄色葡萄球菌菌悬液,离心5分钟,使用pbs缓冲液洗去培养基营养液,该过程重复3次;用pbs缓冲液将离心的菌液稀释到浓度为1×106cfu/ml;将1ml含5.12mmol/lβ-环糊精酰胺化衍生物、1×106cfu/ml金黄色葡萄球菌(s.aureus,阳性菌)的混合液37℃培养3小时,然后使用pbs缓冲液逐步稀释至1×104cfu/ml;取100μl菌液接种到lb琼脂平板上并均匀涂平,37℃孵育24小时,记录菌落数。该实验过程中,每个样品平行测定三次,测定结果见图15。
杀菌率=(cfu空白–cfu样品)/(cfu空白)×100%
从图15中可以看出,β-cd-6-fa对金黄色葡萄球菌杀菌率可以达到28.70%,β-cd-6-p-ca对金黄色葡萄球菌杀菌率可以达到76.86%,β-cd-6-ca对金黄色葡萄球菌杀菌率可以达到84.14%,说明本发明β-环糊精酰胺化衍生物具有良好的抗菌性能。
- 还没有人留言评论。精彩留言会获得点赞!