一种制备广藿香烯酮的方法与流程
本发明属于化学领域,涉及一种制备广藿香烯酮的方法。
背景技术:
广藿香烯酮、香附薁酮、香附子烯-2,5,8-三醇是存在于莎草中的化合物,化学结构如下。
目前制备广藿香烯酮、香附薁酮、香附子烯-2,5,8-三醇的方法比较复杂,需要借助液液萃取、反复硅胶柱层析、葡聚糖凝胶柱层析等手段,难以规模化运用。
技术实现要素:
本发明的目的是提供一种制备广藿香烯酮的方法。
本发明上述目的通过如下技术方案实现:
一种制备广藿香烯酮的方法,包括:
步骤1、乙醇总提物的制备
将莎草的干燥根茎粉碎,用无水乙醇提取,提取液过滤,浓缩干燥得到乙醇总提物;
步骤2、mci微孔树脂富集
将乙醇总提物加水搅拌、超声溶解,过滤,上样于mci微孔树脂柱内,上样的溶液体积为1.5bv,上样流速为1.5bv/h,上样结束后关闭树脂柱阀门,静置吸附0.5h;洗脱时,先用40%乙醇以3bv/h流速洗脱1.5h,再用70%乙醇以3bv/h流速洗脱,收集5~6bv的70%乙醇洗脱液,浓缩干燥得富集物;
步骤3、制备液相色谱分离纯化
将富集物用体积百分浓度为20%的乙醇水溶液溶解配制成进样溶液,过滤,注入制备液相色谱仪进行分离纯化,制备液相色谱参数如下:
色谱柱为ultimatexb-c8(10μm,10mm×250mm),流速为2.5ml/min,柱温25℃,紫外检测波长为225nm,进样量0.5ml;梯度洗脱:流动相a为水:甲酸=100:3,流动相b为乙腈,洗脱程序:0~10min,b为20%;10~25min,b为20%~65%;25~40min,b为65%~100%;
根据色谱图收集广藿香烯酮色谱峰对应的洗脱流份,浓缩干燥即得到广藿香烯酮。
优选地,步骤1用无水乙醇冷浸提取3次,每次24h;提取液过滤,合并,浓缩干燥得到乙醇总提物。
优选地,步骤2将乙醇总提物加水搅拌、超声溶解,配制成浓度为1g/ml的溶液。
优选地,步骤2中mci微孔树脂柱的长径比为6:1。
优选地,步骤3中将富集物用体积百分浓度为20%的乙醇水溶液溶解配制成浓度为10mg/ml的进样溶液。
有益效果:
本发明仅通过mci微孔树脂和反相制备液相色谱即可制备得到纯度在95%以上的广藿香烯酮、香附薁酮、香附子烯-2,5,8-三醇,易于规模化制备。
附图说明
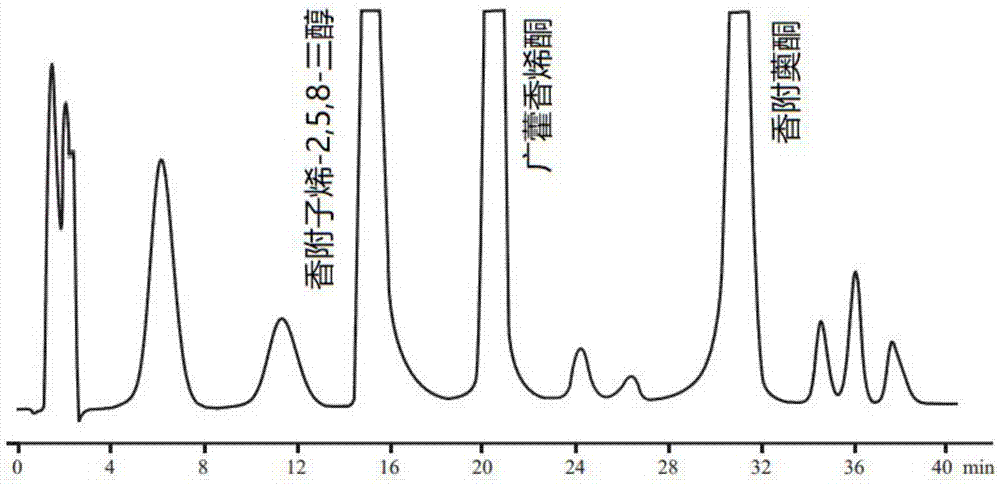
图1为制备液相色谱图;
图2为香附子烯-2,5,8-三醇供试品溶液和对照品溶液色谱图;
图3为广藿香烯酮供试品溶液和对照品溶液色谱图;
图4为香附薁酮供试品溶液和对照品溶液色谱图。
具体实施方式
下面结合实施例具体介绍本发明实质性内容,但并不以此限定本发明的保护范围。
一、实验材料
莎草科植物莎草cyperusrotundusl.的干燥根茎即香附购自安徽亳州中药材市场。
mci柱层析用chp-20p(75~150μm)为三菱化学公司产品。
半制备液相色谱仪为agilent1200,制备柱为ultimatexb-c8(10μm,10mm×250mm),分析柱为agilentzorbaxsbc18(4.6mm×250mm,5μm)。
二、实验方法和结果
1、乙醇总提物的制备
将莎草cyperusrotundusl.的干燥根茎粉碎,用无水乙醇冷浸提取3次,每次24h;提取液过滤,合并,浓缩干燥得到乙醇总提物。
2、mci微孔树脂富集
将乙醇总提物加水搅拌、超声溶解,配制成浓度为1g/ml的溶液,过滤,上样于长径比为6:1的mci微孔树脂柱内,上样的溶液体积为1.5bv,上样流速为1.5bv/h,上样结束后关闭树脂柱阀门,静置吸附0.5h;洗脱时,先用40%乙醇以3bv/h流速洗脱1.5h,再用70%乙醇以3bv/h流速洗脱,收集5~6bv的70%乙醇洗脱液,浓缩干燥得富集物。
3、制备液相色谱分离纯化
将富集物用体积百分浓度为20%的乙醇水溶液溶解配制成浓度为10mg/ml的进样溶液,过滤,注入制备液相色谱仪进行分离纯化,制备液相色谱参数如下:
色谱柱为ultimatexb-c8(10μm,10mm×250mm),流速为2.5ml/min,柱温25℃,紫外检测波长为225nm,进样量0.5ml;梯度洗脱:流动相a为水:甲酸=100:3,流动相b为乙腈,洗脱程序:0~10min,b为20%;10~25min,b为20%~65%;25~40min,b为65%~100%;
根据色谱图(图1)收集香附子烯-2,5,8-三醇、广藿香烯酮、香附薁酮色谱峰对应的洗脱流份,浓缩干燥即得到香附子烯-2,5,8-三醇、广藿香烯酮、香附薁酮。
4、纯度检测
将上述制备得到的香附子烯-2,5,8-三醇、广藿香烯酮、香附薁酮分别用体积百分浓度为20%的乙腈水溶液溶解配制成浓度为1mg/ml的供试品溶液,同时配制浓度为0.1mg/ml的对照品溶液,分别注入液相色谱仪检测,色谱参数如下:
色谱柱为agilentzorbaxsb-c18(4.6mm×250mm,5μm),流速为1ml/min,柱温25℃,紫外检测波长225nm,进样量10μl;梯度洗脱:流动相a为水:乙酸=100:1;流动相b为甲醇:乙腈:乙酸=70:30:1。洗脱程序:0~10min,b为20%;10~40min,b为20%~100%。
香附子烯-2,5,8-三醇、广藿香烯酮、香附薁酮供试品溶液和对照品溶液色谱图如图2~4。
实验结果表明,本发明提供的方法可以制备得到纯度不低于95%的香附子烯-2,5,8-三醇、广藿香烯酮、香附薁酮,方法简易可行,易于规模化制备。
上述实施例用于介绍本发明实质性内容,但并不以此限定本发明的保护范围。
技术特征:
技术总结
本发明公开了一种制备广藿香烯酮的方法,包括:乙醇总提物的制备、MCI微孔树脂富集和制备液相色谱分离纯化。本发明仅通过MCI微孔树脂和反相制备液相色谱即可制备得到纯度在95%以上的广藿香烯酮、香附薁酮、香附子烯‑2,5,8‑三醇,易于规模化制备。
技术研发人员:张长林;杨佳
受保护的技术使用者:淮安培元基因科技有限公司
技术研发日:2018.09.25
技术公布日:2019.02.12
- 还没有人留言评论。精彩留言会获得点赞!