一种基于二代测序数据分析对宏基因组中的沙门氏菌进行鉴定及分型的方法与流程
本发明涉及沙门氏菌检测领域,具体涉及一种基于二代测序数据分析对宏基因组中的沙门氏菌进行鉴定及分型的方法。
背景技术:
随着经济的发展,食品中存在的卫生安全隐患问题愈发严重,沙门氏菌是食品中最常见的致病菌,是导致食物中毒的重要病原菌之一,严重威胁到人类健康和食品安全。据美国食品药品监管局(fda)统计,在美国每年因食源性致病菌感染人数可达4800万,其中住院患者和死亡人数分别为12.8万和3000,造成巨大的经济负担。在我国,每年由沙门氏菌引起的食物中毒事件占到全部食物中毒的40%-60%。
沙门氏菌属肠杆菌科,革兰氏阴性肠道杆菌,目前已经发现1800种以上,按抗原成分可分为甲、乙、丙、丁、戊等基本菌型。其中与人类疾病有关的主要有甲组的副伤寒甲杆菌,乙组的副伤寒乙杆菌和鼠伤寒杆菌,丙组的副伤寒丙杆菌和猪霍乱杆菌,丁组的伤寒和肠炎杆菌。此菌可引起禽伤寒、鸡白痢、猪霍乱、鼠伤寒沙门氏菌病、猪副伤寒、马流产沙门氏菌病等疾病。致病性最强的是猪霍乱沙门氏菌(salmonellacholerae),其次是鼠伤寒沙门氏菌(salmonellatyphimurium)和肠炎沙门氏菌(salmonellaenteritidis)。
目前,在我国普遍采用传统的细菌学检测方法和血清学方法,这些检测方法大致需要4到6天才能得到有效的结果,具有检测时间长、操作繁琐的缺陷,难以应对突发疫情的发生。
有鉴于此,本发明人针对上述对宏基因组中的沙门氏菌进行鉴定及分型的方法上未臻完善所导致的诸多缺失及不便,而深入构思,且积极研究改良试做而开发设计出本发明。
技术实现要素:
本发明的目的在于提供一种基于二代测序数据分析对宏基因组中的沙门氏菌进行鉴定及分型的方法,其检测快速而精确。
为实现上述目的,本发明采用的技术方案是:
一种基于二代测序数据分析对宏基因组中的沙门氏菌进行鉴定及分型的方法,其包括以下步骤:
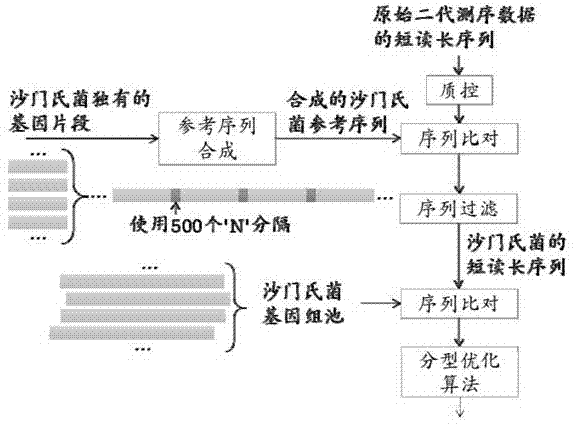
步骤1、构建沙门氏菌的参考序列以及基因组池
步骤1.1、以沙门氏菌独有的基因组序列区域依次首尾连接构建一条参考序列,该参考序列用于短序列过滤;在参考序列中,每相邻两段独有的基因组序列之间采用n连接;
步骤1.2、构建沙门氏菌的基因组池,该基因组池中包含了所有沙门氏菌的基因组序列;
步骤2、对检测样本中的沙门氏菌进行鉴定及分型
步骤2.1、从检测样本中筛选出沙门氏菌;
首先,对检测样本中的宏基因组进行二代测序,得到样本中宏基因组的二代测序数据;
然后,将宏基因组的二代测序数据与步骤1.1中构建的沙门氏菌的参考序列进行比对,筛除非沙门氏菌独有的短读长序列,并且得到对比后的短读长序列;
步骤2.2、根据步骤2.1筛选出来的沙门氏菌独有的短读长序列,通过与沙门氏菌基因组池里面的所有沙门氏菌基因组序列进行比对,对检测样本中的沙门氏菌进行分型。
所述步骤2.1中,将宏基因组的二代测序数据与参考序列进行比对时,进行以下过滤:
(1)对于双端二代测序数据,过滤掉一端没有比对上、或者两端均没有比对上的短读长序列,仅留下两端均成功比对上的数据;
(2)对于两端均成功比对上的二代测序数据,过滤掉满足以下三个条件之一的数据:
(a)短读长序列与比对上的参考序列的距离nm>5;
(b)比对结果的剪切长度clipping>10;
(c)短读长序列本身的读长readlength<100。
所述步骤1.2中,构建基因组池的方法具体如下:
从网上公开数据库下载所有沙门氏菌的基因组序列和测序原始数据,对于完整的基因组序列,过滤掉质粒部分序列,得到只含有沙门氏菌的基因组序列;对于有很多个重叠群组成的基因组序列,将重叠群序列用n连接,形成沙门氏菌的参考基因组序列;对于沙门氏菌的测序原始数据,进行组合形成沙门氏菌的基因组序列;将所述沙门氏菌的基因组序列以及参考基因组序列放入基因组池中形成沙门氏菌的基因组池。
所述步骤2用如下办法进行分型:
首先,将步骤2.1筛选出来的短读长序列与基因组池中的所有沙门氏菌的基因组序列进行比对,得到对比结果;
然后,采用最小覆盖优化算法msc(minimumsetcover)对上述比对结果进行优化处理;优化方程为:
c(i)=(|∪i∈i5i|-γ|i|)
r={rj|j=1,…,j}
其中,i代表检测样本中含有的沙门氏菌短读长序列的集合;
u代表基因组池中所有沙门氏菌的基因组序列;
r代表过滤后的短读长序列的集合;
│·│代表集合的基数;
γ代表稀疏调节参数,用于控制最优解的稀疏度,γ越大代表能获得的最优解的数据集就越小。
所述步骤2.2中,优化方程采用贪心算法求解。
采用本发明的技术方案后,本发明首先构建了沙门氏菌的参考序列以及沙门氏菌的基因组池,在对检测样本中的沙门氏菌进行鉴定及分型时,将检测样本的二代测序数据与沙门氏菌的参考序列进行比对,筛除非沙门氏菌的短读长序列,得到过滤后的短读长序列;再将该过滤后的短读长序列与基因组池中的所有沙门氏菌的基因组序列进行比对,并进行优化处理,得到优化结果用以确定沙门氏菌及其类型。该方法具有检测快速而精确的优点,首先,采用沙门氏菌独有的基因片段,合成为一条特定的参考基因序列,通过将短读长序列比对到该参考基因序列,实现序列过滤,去除了不相关物种的短读长序列,同时降低了后续比对及优化算法的复杂度。其次,过滤后的短读长序列再通过序列比对的办法,比对到参考基因组数据库,通过独有的优化算法分析比对结果从而实现菌群的定株。由于非目标菌株的短读长序列已经在去噪步骤被清除,该序列比对步骤精确度高而且计算复杂度低。
附图说明
图1为本发明沙门氏菌的的鉴定及分型方法流程图。
具体实施方式
如图1所示,本发明揭示了一种基于二代测序数据分析对宏基因组中的沙门氏菌进行鉴定及分型的方法,其包括以下步骤:
步骤1、构建沙门氏菌的参考序列以及基因组池;
步骤1.1、以沙门氏菌独有的基因组序列区域依次首尾连接构建一条参考序列,该参考序列用于短序列过滤。参考序列的构建具体如下:
沙门氏菌共有403段独有的基因组序列区域,每段独有的基因组序列区域含有1000碱基(bp),提取上述403段沙门氏菌独有的基因组序列区域,并且以该独有的基因组序列区域依次首尾连接合成一条参考序列。
在参考序列中,每相邻两段独有的基因组序列区域之间采用500个n字符连接,从而避免在比对时发生样本的短读长序列比对到相邻两段独有的基因组序列区域的首尾连接的位置,导致出现错误的比对结果。当然,n的数量也不仅仅限于500。
步骤1.2、构建沙门氏菌的基因组池,该基因组池中包含了所有沙门氏菌的基因组序列。
从网上公开数据库下载所有沙门氏菌的基因组序列和测序原始数据,对于完整的基因组序列,过滤掉质粒部分序列,得到只含有沙门氏菌的基因组序列;对于有很多个重叠群组成的基因组序列,将重叠群序列用一定数量的n字符连接,形成沙门氏菌的参考基因组序列;对于沙门氏菌的测序原始数据,进行组合形成沙门氏菌的基因组序列。将上述沙门氏菌的基因组序列以及参考基因组序列放入基因组池中形成沙门氏菌的基因组池。例如,可以从以下网站上下载,构建基因组池序列:
(1)从ncbi网站下载所有沙门氏菌菌株完整的基因组和染色体序列,并过滤掉质粒部分序列,得到只含有沙门氏菌的基因组序列。
(2)从sistr网站下载所有的沙门氏菌基因组。这些基因组序列不是完整序列,而是由很多个重叠群(contig)组成。与步骤1.1中合成沙门氏菌特有的参考序列的方法类似,将这些重叠群序列用不短于500个n连接,做为参考基因组序列,放入沙门氏菌基因组池。
(3)除此以外,从ncbi网站下载沙门氏菌的测序原始数据(fastq文件),用spades软件组装成沙门氏菌基因组序列。
沙门氏菌基因组池里的沙门氏菌基因组序列(fasta文件)通常都在文件头包含有其菌株名和血清型。对于基因组池中不包含mlst分型的沙门氏菌,可以采用stringmlst软件得到其mlst分型。
步骤2、对检测样本中的沙门氏菌进行鉴定及分型
步骤2.1、从检测样本中筛选出沙门氏菌
bwa是一款基于bwt的快速比对工具,其由三个算法组成。这三个算法分别是:bwabacktrack,bwaswandbwamem。
首先,对样本进行二代测序,得到样本的二代测序数据。然后对样本的二代测序数据进行质控和预处理,以保证二代测序数据的干净可靠。
然后,使用bwamem将宏基因组的二代测序数据与步骤1.1中构建的沙门氏菌的参考序列进行比对,筛除非沙门氏菌的短读长序列,并且得到对比后的短读长序列。
将宏基因组的二代测序数据与参考序列进行比对时,进行以下过滤,以保证对比得到的短读长序列属于沙门氏菌对双端测序数据。过滤条件如下:
(1)对于双端二代测序数据,过滤掉一端没有比对上、或者两端均没有比对上的短读长序列,仅留下两端均成功比对上的数据,该过滤可以用samtools软件完成;
(2)对于两端均成功比对上的二代测序数据,过滤掉满足以下三个条件之一的数据:
(a)短读长序列与比对上的参考序列的距离nm>5;
(b)比对结果的剪切(软剪切或硬剪切)长度clipping>10;
(c)短读长序列本身的读长readlength<100。
步骤2.2、根据步骤2.1筛选出来的短读长序列对检测样本中的沙门氏菌进行分型
通过bwamem将筛选出来的短读长序列与基因组池中的所有沙门氏菌的每一条基因组序列分别进行比对。对每一条短读长序列,可以通过比对得到该基因组池的一个子集,该短读长序列能够正确比对到该子集里的每一条基因组序列上,而不能比对到该子集的补集里的任何一条基因组序列上。
由于沙门氏菌的不同菌株的基因组之间极其相似,在短读长序列与基因组池的比对中,很多同种沙门氏菌能够比对到不同的沙门氏菌菌株上。为了能够得到最好的鉴定结果,本发明采用基于最小覆盖优化算法msc(minimumsetcover)对上述比对结果进行优化处理。该优化处理的目标是针对输入的短读长序列数据,从沙门氏菌的基因组池里找到一个最小的参考序列的子集,可以提供最佳覆盖。优化方程如下所示:
c(i)=(|∪i∈isi|-γ|i|)
r={rj|j=1,…,j}
其中,i代表检测样本中含有的沙门氏菌短读长序列的集合;
u代表基因组池中所有沙门氏菌的基因组序列;
r代表(根据步骤2.1)过滤后的短读长序列的集合;
│·│代表集合的基数;
γ代表稀疏调节参数,用于控制最优解的稀疏度,γ越大代表能获得的最优解的数据集就越小。
对上述优化方程进行求解,即可得到检测样本中含有的沙门氏菌的集合i,并得到该集合i中的沙门氏菌的类型。
上述优化方程可以采用如下贪心解法来求解,具体如下:
1、i←φ,其中,φ为空集合;
2、计算覆盖强度权重;
3、对每个沙门氏菌
4、选择具有最高覆盖权重的沙门氏菌i0;
5、如果c(i)<c(i+i0),i←i+i0,返回步骤2;
6、否则,输出i。
当然,上述算法只是优化方程的解法之一,也可以使用其他算法对优化方程进行求解。
本发明的关键在于,本发明首先构建了沙门氏菌的参考序列以及沙门氏菌的基因组池,在对检测样本中的沙门氏菌进行鉴定及分型时,将检测样本的二代测序数据与沙门氏菌的参考序列进行比对,筛除非沙门氏菌的短读长序列,得到过滤后的短读长序列;再将该过滤后的短读长序列与基因组池中的所有沙门氏菌的基因组序列进行比对,并进行优化处理,得到优化结果用以确定沙门氏菌及其类型。首先,本发明的去噪方案通过序列过滤,去除了不相关物种的短读长序列,减少了噪声数据对后续比对及优化算法的干扰,同时,因为滤除过的短读长序列比较少,这个办法同时也降低了后续操作的复杂度。最后,通过贪心算法求解优化问题,计算复杂度低,运行速度快。简言之,本发明的方法具有检测快速而精确的优点。
以上所述,仅是本发明实施例而已,并非对本发明的技术范围作任何限制,故凡是依据本发明的技术实质对以上实施例所作的任何细微修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!