苯并咪唑并咪唑衍生物及其合成方法与流程
本发明涉及苯并咪唑并咪唑衍生物及其合成方法,特别涉及一种通过溶剂热一锅法合成苯并咪唑并咪唑衍生物的方法。
背景技术:
苯并咪唑并咪唑及其衍生物在稻瘟病防治、生物医药以及配合物发光方面有潜在应用,因此受到了广泛的关注。传统合成方法先合成酰胺中间体,再分子内关环,而且需要使用对环境不友好的pocl3等试剂,从而限制了这类化合物的合成。
溶剂热反应即将简单原料及溶剂在反应釜中密闭,置于适宜的温度下进行反应。近年来该反应被广泛用于合成常规方法难以得到的结构新颖的化合物,这些化合物通常在光、电、磁、催化以及生物上有潜在应用。值得注意的是,溶剂热条件下,配体有时会发生原位反应,形成新的有机物,因此,溶剂热反应为合成具有特定功能的复杂分子提供了新的方法。
技术实现要素:
本发明的目的是提供一类结构新颖的苯并咪唑并咪唑衍生物;
本发明的另一个目的是提供上述苯并咪唑并咪唑衍生物的制备方法;
本发明实现过程如下:
结构通式l2所示化合物,
r1选自氢原子、c1~c6的烷基、c1~c6的烷氧基、苯基、氰基、三氟甲基、卤素基;
r2选自苯基、环已基、c1~c6的烷基。
上述化合物的制备方法:
将化合物l1溶于异丙醇中,加入催化量的六水合三氯化铁,在80~140℃条件下密闭反应得到化合物l2。
本发明利用l1(如苯并咪唑甲胺)为原料,在fecl3∙6h2o催化下,以异丙醇为溶剂,通过溶剂热反应一锅法得到目标化合物苯并咪唑并咪唑衍生物l2,另外,利用电喷雾质谱对反应的溶剂及反应时间进行了筛选,确定了fecl3·6h2o催化剂的重要作用。在此基础上,提出了可能的反应机理,为合成这类型的含氮杂环化合物提供了简便且环境友好的合成方法。
附图说明
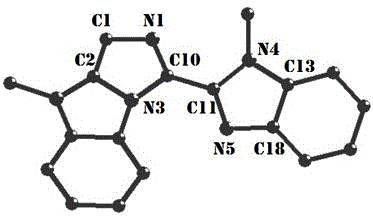
图1为化合物l2的单晶结构图;
图2为化合物l2的核磁氢谱、碳谱及dept-135谱;
图3为化合物l2的电喷雾离子化高分辨质谱图及质谱片段拟合;
图4为化合物l2的热重分析图;
图5为化合物l2的红外图;
图6为化合物l2的紫外光谱吸收图;
图7为化合物l2的荧光发射谱图;
图8为l1和fecl3∙6h2o在不同醇溶剂中反应的质谱图;
图9为l1和fecl3∙6h2o在异丙醇中反应不同时间的质谱图;
图10为fecl3∙6h2o对反应影响的质谱图;
图11为化合物l2可能的形成机理。
具体实施方式
为了更清楚的理解发明,以下通过发明人给出的依本发明技术方案所完成的具体实施例对本发明进一步的详细描述。
实施例1:l2(r1=h,r2=ch3)的合成及结构分析
将1.0mmol的n-甲基苯并咪唑甲胺(161.0mg)加入到容积为100.0ml的聚四氟乙烯的反应釜中,加入40.0ml的异丙醇溶解后,再加入0.2mmol的六水合三氯化铁(54.0mg),搅拌5分钟,密封,放入140℃的微波消解仪中反应10min,然后将母液旋干、制样并进行柱层析分离,产物通过界面扩散得到浅黄色单晶,其属于单斜晶系,p21空间群,晶胞参数为a=5.4720(1)å,b=8.9105(2)å,c=15.3895(4)å,α=90o,β=99.801(2)o,γ=90o。核磁数据也验证了该单晶结构:1hnmr(cd2cl2,500mhz)9.45(d,j=10.0hz,1harh),7.94-7.91(m,1h,arh),7.44-7.42(m,2h,arh),7.37-7.55(m,2h,arh),7.30-7.27(m,1h,arh),7.23(d,j=10.0hz,1h,arh),6.84(s,1h),4.37(s,3h,ch3),3.75(s,3h,ch3).13c{1h}nmr(cd2cl2,125mhz):144.2,142.9,142.7,139.2,136.6,125.9,125.2,122.6,122.2,119.7,119.4,117.5,109.3,108.2,98.9,77.2,32.1,30.5。
产物表征测试:
图1为l2的单晶结构图,结果表明l2是由苯并咪唑及苯并咪唑并咪唑两个含氮杂环构成,二面角为13.3o,表明l2的结构没有完全共平面;
图2为l2的核磁氢谱和碳谱以及dept-135谱。室温下,在bruker500mhz上进行l2的核磁共振谱图测试,取15.0mg的l2溶于0.5ml的cd2cl2中进行测试,得到的谱图结果与单晶结构相吻合;
图3为l2的电喷雾离子化高分辨质谱图,室温下,取1颗单晶溶于色谱级乙腈中,在thermoexactiveplusesi-ms上进行阳离子模式下的数据采集,m/z在302.1406处的峰为分子离子峰[l2+h]+;
图4为化合物l2的热重分析图,室温下称取6mg的l2,在netzschtg209f3上进行l2热稳定测试,其中加热区间为25-800oc,升温速率为5oc/min,氮气流速为15ml/min,结果表明氮气气氛下l2在220℃开始分解;
图5为化合物l2的红外图。室温下,在brukerftirspectrophotometer上进行l2的红外测试,取0.1mg的l2制成kbr压片,测试范围为4000-400cm-1;
图6为化合物l2的紫外光谱吸收图。室温下,在agilentcary6000uv-vis-nirspectrophotometer上对l2的乙腈溶液(10-5mol•l-1)进行测试。如图6所示,l2的乙腈溶液在296nm和380nm有最大吸收峰,可以归属为π-π*跃迁;
图7为化合物l2的荧光发射谱图。室温下在爱丁堡fls-980荧光仪上对l2的乙腈溶液(10-5mol•l-1)进行测试。如图7所示,在波长为249nm光激发下,在可见-近红外(370-480nm)具有荧光发射,其中434nm为分子内的跃迁,510nm为分子间的π-π堆积;
图8为l2和fecl3∙6h2o在不同醇溶剂中反应的质谱图,说明异丙醇溶剂的使用对最终产物的合成起到了非常特殊的作用;
图9为l2和fecl3∙6h2o在异丙醇中反应不同时间的质谱图;
图10为fecl3∙6h2o对反应影响的质谱图;说明催化剂fecl3∙6h2o是必不可少的;
图11为化合物l2可能的形成机理。根据文献,初步推测了可能的反应机理:首先配体l1与fecl3配位,接着配体发生氧化自缩合得到亚胺中间体,然后发生分子内亲核加成/关环,再脱氢得到l2的fe配合物,最后发生解离得到l2。
实施例2:本发明涉及的结构类似的化合物
参照实施例1,使用不同的原料,制备苯并咪唑衍生物,其结果如下表所示。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种制备1,2?二甲基咪唑的方法
- 一种芬苯达唑纳米混悬剂及其制备方法
- 吡啶并咪唑类衍生物、其制备方法和应用
- 一种咪唑类衍生物化合物及其制备方法和发光器件的制作方法
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗红细胞低下性贫血药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗骨质疏松药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗急性痛风药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在预防或治疗胰腺纤维化药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗缺氧药物中的应用
- 适用于改进水分管理且提供抗微生物保护的包括衍生自直链四胺的季双咪唑啉化合物的 ...的制作方法