低分子量透明质酸的制备方法与流程
本发明涉及一种具有优异的皮肤渗透和保湿效果的分子量范围的透明质酸的制备方法。
背景技术:
透明质酸基于d-葡糖醛酸和n-乙酰-d-葡糖胺,分子量为数十万至数百万道尔顿,几乎所有具有相同结构的生物体中发现的线性聚合物,并且是细胞外基质的组成部分。由于没有物种之间的差异具有相同的结构,因此,无论来源如何都没有免疫反应。迄今为止是一种用于退行性关节炎、白内障、皱纹改善、给药、干细胞支持和化妆品保湿成分等领域的安全的天然多糖。作为聚合物的透明质酸具有密度变高,分子量大,粘度高的特征。
最初,透明质酸是从鸡冠中提取的,并且在工业上用于医药、化妆品和食品【biomacromolecules,5,2122-2127(2004)】。然而,最近,由于担心可能由禽流感等动物引起的传染病,主要通过使用微生物的发酵来产生。【申请微生物。生物技术。66(4):341-51(2005)】。
据报道,人体皮肤中透明质酸的量随着衰老而降低,这被认为是皮肤弹性和含水量降低的直接原因之一。【in:freeradicaldamageantitscontrol,281-300,elsevier科学(1994年)】。
透明质酸因其能够溶于水并在其分子结构中保持大量水分而被广泛用作化妆品的保湿剂,适用于火伤或创伤的乳霜、注射剂、纱布等,也用作保护角膜的内侧的配方。此外,透明质酸已经用于组织工程领域,用于生产用于伤口涂层和牙科基质的多孔海绵状基质。
然而,通过从动物组织中提取或发酵产生的透明质酸通常是500,000道尔顿或更高的聚合物多糖,具有高聚合度,因此通常不会渗透皮肤。即,当将500,000道尔顿以上的透明质酸应用于皮肤时,由于吸收空气中水分的性质,可以防止皮肤水分蒸发,但由于不会渗透皮肤,会停留在皮肤表面,很容易被洗脸时从皮肤表面洗掉,因此,皮肤的保湿效果难以维持。另一方面,100,000道尔顿以下的透明质酸可以渗透皮肤,但分子尺寸小,因此缺乏含水分的性质不能持久保湿效果。
此外,20,000道尔顿以下的ha有效地穿透皮肤,但已知会引起促炎反应,这是因为免疫反应可以通过结合皮肤巨噬细胞上的toll样受体2和4来启动。
低分子量透明质酸的实例包括γ射线,超声波或紫外线(journalofchromatography,435,pp335-342,1988;int.biol.macrmol。,p10,1988),使用酸或碱催化剂的方法(日本专利公开号s63-57602),使用微流化器的方法等是公知的。然而,公知的方法是制备100,000道尔顿以下的透明质酸的方法,并且没有给予皮肤渗透的直接效果或有效制备能够吸收和含有水分的分子量范围内的透明质酸的方法。
技术实现要素:
要解决的技术问题
本发明是鉴于所述诸多问题而提出的,其目的在于,提供一种在保持用作现有化妆品原料的超过500,000道尔顿的透明质酸的保湿效果的同时,克服了不渗透到皮肤角质层中并被洗清不能长时间提供皮肤保湿效果的缺点,渗透到皮肤并且能够保持最佳的湿润状态的有效制备低分子量透明质酸的方法。
技术方案
为了实现所述目的,用于解决本发明中的上述问题的手段如下。
1.一种低分子量透明质酸的制备方法,其中,分子量为100,000至200,000道尔顿,并且包括在2.5至3.5的ph下热处理含有分子量为500,000道尔顿以上的透明质酸的水溶液。
2.根据权利要求1所述的低分子量透明质酸的制备方法,其中,所述水溶液的浓度为1至2%(重量/体积)。
3.根据权利要求1所述的低分子量透明质酸的制备方法,其中,所述热处理温度为80至90℃。
4.根据权利要求1所述的低分子量透明质酸的制备方法,其中,所述热处理时间为15分钟至30分钟。
5.根据权利要求1所述的低分子量透明质酸的制备方法,其中,在所述热处理后,用碱金属氢氧化物水溶液中和所述反应溶液,获得碱金属盐形态的低分子量透明质酸。
6.根据权利要求5所述的低分子量透明质酸的制备方法,其中,所述碱金属盐为钠盐。
7.根据权利要求5所述的低分子量透明质酸的制备方法,其中,在中和后,在反应溶液中添加有机溶剂,形成沉淀,过滤后,得到粉末状的透明质酸的碱金属盐。
8.根据权利要求7所述的低分子量透明质酸的制备方法,其中,所述有机溶剂是选自甲醇、乙醇、丙酮和异丙醇中的一种以上。
9.根据权利要求7所述的低分子量透明质酸的制备方法,其中,基于所述反应溶液,以1∶5至1∶6的体积比添加所述有机溶剂。
有益效果
根据本发明,通过省略各种现有的复杂方法,通过简化的方法克服现有的作为化妆品原料的透明质酸的缺点的难以渗透皮肤的问题,除了皮肤渗透之外,还提供了一种制备分子量为100至200kda的能够保持最大保湿能力的透明质酸的方法。通过本发明提出的方法制备的低分子量透明质酸可渗透皮肤并保持最大水分,因此,缓和过敏症,从而适合化妆品,医药品及医药组合物。
附图说明
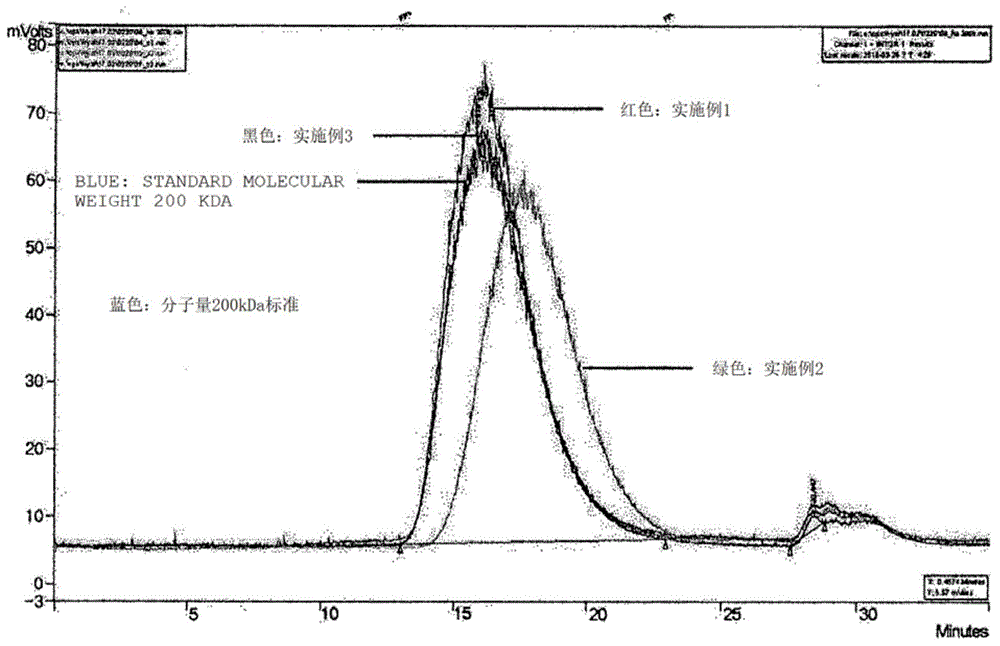
图1是hplc色谱图,证实了实施例1至3中制备的低分子量透明质酸钠的分子量分布。(蓝色:分子量200,000道尔顿标准,红色:实施例1,绿色:实施例2,黑色:实施例3)
图2是hplc色谱图,证实了在本发明的实施例4和5中制备的低分子量透明质酸钠的分子量分布。(蓝色:分子量200,000道尔顿标准,红色:实施例4,绿色:实施例5)
图3是hplc色谱图,证实了在本发明的实施例6和7中制备的低分子量透明质酸钠的分子量分布。(蓝色:分子量200,000道尔顿标准,红色:实施例6,绿色:实施例7)
图4是hplc色谱图,证实了比较例1至3中制备的透明质酸钠的分子量分布。(蓝色:分子量200,000道尔顿标准,红色:分子量100,000道尔顿标准,绿色:比较例1,黑色:比较例2,粉红色:比较例3))
图5是hplc色谱图,证实了在本发明的比较例4至6中制备的透明质酸钠的分子量分布。(蓝色:分子量200,000道尔顿标准,红色:分子量100,000道尔顿标准,绿色:比较例4,黑色:比较例5,粉红色:比较例6))
图6是将荧光标记的本发明的实施例4,对照组和比较例7的样品在balb/c-裸鼠背部涂布10μl(10μg/ml)后,15分钟后用pbs缓冲液清洗老鼠的左侧,30分钟后清洗右侧后去除残留物,然后经过30分钟后,1小时后,2小时后,6小时后,24小时后立即用ivissepctrum测量。
图7是将荧光标记的本发明的实施例4,对照组和比较例7的样品在balb/c-裸鼠背部涂布10μl(10μg/ml)后,15分钟后用pbs缓冲液清洗去除残留物后,取出小鼠的皮肤切片,用4%甲醛固定以制备切口标本,并使用双光子显微镜对皮肤组织切片进行共焦成像。
具体实施方式
本发明涉及分子量为100,000至200,000道尔顿的低分子量透明质酸的制备方法,该方法包括在2.5至3.5的ph下热处理含有分子量为500,000道尔顿以上的透明质酸的水溶液。
在本发明中,用作起始材料的分子量为500,000至3,000,000道尔顿的透明质酸包括一般的透明质酸化妆品级原料为500,000道尔顿的平均分子量,100万道尔顿的药物原料级和300万道尔顿的透明质酸。
通常,优选使用价格低廉的化妆品原料平均分子量为500,000道尔顿的透明质酸,但不限于此。
所述水溶液可以是溶解在水中的分子量为500,000至3,000,000道尔顿透明质酸的溶液,优选地浓度为1至2%(重量/体积)的蒸馏水,所述酸是盐酸、磷酸、乙酸等本发明领域中通常使用的酸中的任一个。
所述热处理温度优选地80至90℃以提高工程效率性,热处理时间取决于原料优选为15至30分钟。
在本发明中,通过将含有分子量为500,000道尔顿以上的透明质酸的水溶液调节至2.5至3.5的ph并在相同的温度和时间下进行热处理,可以获得平均分子量为100,000至200,000道尔顿的透明质酸。
在本发明中,在所述热处理之后,可以用碱,优选碱金属氢氧化物水溶液中和反应溶液,以获得碱金属盐形态的低分子量透明质酸。在这种情况下,将反应溶液的ph调节至6.5以上,优选6.5至7.0。所述碱金属氢氧化物可以是氢氧化钠,在这种情况下,可以获得低分子量透明质酸的钠盐。
通过上述方法制备的低分子量透明质酸的金属盐可以通过添加有机溶剂加入到反应溶液中以形成沉淀物并过滤以回收粉末形态的低分子量透明质酸的碱金属盐。所述有机溶剂可以是选自甲醇、乙醇、丙酮和异丙醇中的一种以上,并且可以基于反应溶液以1∶5至1∶6的体积比加入。
实施例
在下文中,将参考实施例更详细地描述本发明。然而,下面给出的实施例仅是用于实施本发明的实施例,并且本发明的范围不限于这些实施例的范围。
1.优化低分子量透明质酸钠的制备条件
实施例1
将市场上销售的化妆品级透明质酸钠(bloomagefredabiopharm。产品,分子量500,000至120,0000道尔顿)1g溶解在100ml蒸馏水中以制备1.0%(w/v)水溶液,并加入0.58ml的4nhcl调节至ph2.5,调节后,将在90℃的水中浸泡15分钟。然后,将混合物放入冷水中以将温度降至室温,向其中加入4nnaoh溶液以将ph调节至6.5,并加入400ml无水乙醇以形成沉淀物。滤出沉淀物并在50℃下干燥,得到8.9g透明质酸钠。
实施例2
以与实施例1相同的方式获得8.7g透明质酸钠盐,不同之处在于将混合物在90℃的水中加热30分钟。
实施例3
以与实施例1相同的方式获得9.1g透明质酸钠盐,不同之处在于,将0.23ml的4nhcl溶液加入透明质酸钠水溶液中,调节至ph3.5并在90℃下在水中加热30分钟。
图1示出实施例1(红色峰)和实施例3(黑色峰)中获得的透明质酸钠盐的平均分子量与200,000道尔顿标准品(蓝色峰)重叠,并且实施例2中(绿色峰)获得的透明质酸钠盐的分子量小于200,000道尔顿(蓝色峰)。
实施例4
以与实施例1相同的方式获得8.9g透明质酸钠盐,不同之处在于向透明质酸钠水溶液中加入0.21ml4nhcl溶液以将ph调节至3.0。
实施例5
以与实施例1相同的方式获得8.2g透明质酸钠盐,不同之处在于向透明质酸钠水溶液中加入0.21ml4nhcl溶液以将ph调节至3.0。
图2显示实施例4(红色峰)中得到的透明质酸钠盐的平均分子量与200,000道尔顿标准重叠,实施例5(绿色峰)中得到的透明质酸钠盐的分子量小于200,000道尔顿标准(蓝色峰)。
实施例6
以与实施例1相同的方式获得8.7g透明质酸钠盐,不同之处在于向透明质酸钠水溶液中加入0.21ml4nhcl溶液以将ph调节至3.0并在80℃下在水中加热15分钟。
实施例7
以与实施例1相同的方式获得8.5g透明质酸钠盐,不同之处在于向透明质酸钠水溶液中加入0.21ml4nhcl溶液以将ph调节至3.0并在80℃下在水中加热30分钟。
图3显示实施例6(红色峰)中得到的透明质酸钠盐的分子量分布几乎与200,000道尔顿标准(蓝色峰)一致,结果表明,实施例7(绿色峰)中得到的透明质酸钠盐的平均分子量小于200,000道尔顿标准(蓝色峰)。
比较例1
将市场上销售的化妆品级透明质酸钠(bloomagefredabiopharm。产品,分子量500,000至120,0000道尔顿)1g溶解在100ml蒸馏水中以制备1.0%(w/v)水溶液,并加入0.62ml的4nhcl调节至ph2.0,调节后,将在90℃的水中浸泡15分钟。然后,将混合物放入冷水中以将温度降至室温,向其中加入4nna0h溶液以将ph调节至6.5,并加入400ml无水乙醇以形成沉淀物。滤出沉淀物并在50℃下干燥,得到7.3g透明质酸钠。
比较例2
以与实施例1相同的方式获得6.9g透明质酸钠盐,不同之处在于将混合物在90℃的水中加热30分钟。
比较例3
以与实施例1相同的方式获得9.3g透明质酸钠盐,不同之处在于,将0.18ml的4nhcl溶液加入透明质酸钠水溶液中,调节至ph4.0并在90℃下在水中加热30分钟。
图4显示比较例1(绿色峰),比较例2(黑色峰)和比较例3(粉红色峰)中获得的透明质酸钠盐的分子量小于100,000道尔顿标准(红色峰)。
比较例4
以与实施例1相同的方式获得9.1g透明质酸钠盐,不同之处在于向透明质酸钠水溶液中加入0.21ml4nhcl溶液以将ph调节至3.0并在70℃下在水中加热30分钟。
比较例5
以与实施例1相同的方式获得7.8g透明质酸钠盐,不同之处在于向透明质酸钠水溶液中加入0.21ml4nhcl溶液以将ph调节至3.0并在100℃下在水中加热15分钟。
比较例6
以与比较例5相同的方式获得7.4g透明质酸钠盐,不同之处在于在100℃下在水中加热30分钟。
图5显示比较例4(绿色峰),比较例5(黑色峰)和比较例6(粉红色峰)中获得的透明质酸钠盐的分子量小于100,000道尔顿标准(红色峰值)。
实验例
实施例1.使用fff-mals确认分子量分布
所述实施例4中制备的透明质酸钠盐及比较例7(市售化妆品级透明质酸钠(bloomagefredabiopharm。产品,分子量500,000至1.20万道尔顿)分别溶解在蒸馏水中以制备1.0%(w/v)水溶液作为分析样品,通过fff/mals(流场-流动分数/多角度光散射)分析仪(wyatttechnology)分析分子量分布。结果如下表1所示。
分析条件如下。
垫片厚度:250μm
膜:复合再生纤维素20kda(millipore)
注射量:20μg
样品浓度:1毫克/毫升
载体溶液:0.1mnano3+0.02%nan3
样品流速:0.1ml/min
交叉流速:2.0ml/min,0-4分钟;0.5ml/min,持续4-5分钟;0.1毫升/分钟,5-6分钟;0.02毫升/分钟,持续6-8分钟
如表1所示,在本发明的实施例4中制备的透明质酸钠盐的平均分子量为160,000道尔顿,将一般化妆品原料(比较例7)分析为平均分子量为1百万道尔顿。
实验例2.使用hplc确认分子量
使用hplc分析仪(varianprostar210溶剂输送系统)确认实施例1至比较例7和比较例1至6中制备的透明质酸盐的分子量。将10mg所述实施例1至7和比较例1至6中制备的透明质酸盐样品和参考物质(lifecore的100kda和200kda产品)称重并溶解在10ml流动相中,并且用0.45μm过滤器过滤后,然后进行分析。结果显示在图1-6中。
分析条件如下
柱:1)ultrahydrogel1000,12μm,7.8×300mm,2k-4m柱/2)ultrahydrogel500,10μm,7.8×300mm,10k-400k柱,串联连接)
柱温箱:室温
流速:0.8毫升/分钟
注射体积:100μl
探测器:alltechelsd3300
气体流量:1升/分钟,温度:70℃
流动相:20mm-碳酸铵(ph7.8)
分子量参考:200kda,100kdaha
实验例3.通过测定特性粘度测定水含量
为了根据分子量确认水含量,根据测量欧洲药典的透明质酸钠的特性粘度的方法进行试验。
作为粘度计,使用乌氏粘度计型粘度计。将实施例4和5中制备的透明质酸钠盐比较例7及分别来自参考物质(lifecore的分子量为20kda,10kda,5kda的透明质酸)(m0p)分别加入10.0g(m0s)缓冲溶液中,将混合物在4℃下振荡24小时,通过玻璃过滤器过滤,并用作样品溶液(t0)。将50g缓冲溶液加入到10g(t0)样品溶液中,然后在25℃下振荡20分钟,然后通过玻璃滤器过滤。弃去5ml第一滤液,下一滤液用作样品溶液t1。将5g缓冲溶液加入到15g测试溶液(t1)中,并以与上述相同的方式制备测试溶液t2。将10g缓冲溶液加入到10g测试溶液(t1)中,并以与上述相同的方式制备测试溶液t3。将15g缓冲溶液加入到5g测试溶液(t1)中,得到试测试溶液t4。
将样品溶液放入粘度计中,使液位在球体a上的两个标记之间。将粘度计置于由兽药规定的温度(0.1℃)下的恒温器中,使球d完全浸没并垂直保持,并静置约20分钟直至样品溶液达到指定温度。用手指挡住管子m,从管子n的上端轻轻吸入,防止气泡进入管子n,将液面拉到球体d的中心,停止抽吸,打开管子m的入口,立即堵住管子m的入口。在确认毛细管最下端的液柱破裂后,打开管子n的入口并测量液面从球c的上线流到下线的时间t(秒)。
1.测量粘度法:毛细管粘度计法
作为测量牛顿液体粘度的方法,测量一定体积的液体流过毛细管所需的时间t(s),并且根据下面的等式(1)计算动力粘度ν。
【等式1】
v=k·t
为了求粘度η,再次测量该温度下液体的密度ρ(g/ml)并根据下式2计算。
【等式2】
η=ν·ρ=k·t·ρ
在等式1和等式2中,k(mm2/s2)是粘度计的整数,并且使用用于粘度计校准的标准溶液预先确定。特性粘度表示聚合物在液体(样品液体)中的扩散程度,并且可以是分子量的标准。测量具有浓度c(g/dl)的样品液体流下的时间t和溶剂流下的时间to,并且根据下等式3计算极限粘度【η】。
2.缓冲溶液:在ph7.0的磷酸钠缓冲溶液中的0.15m氯化钠
溶液a:通过将1.56gnah2po4和9.0gnacl溶解在1l蒸馏水中制备
溶液b:通过将3.58gna2hpo4和9.0gnacl溶解在1l蒸馏水中制备。
将溶液a与溶液b混合至ph7.0并用玻璃滤器过滤
3.验证实验
测量测试溶液t1,t2,t3,t4和缓冲溶液在25℃下沿毛细管粘度计流动的时间分别为t1,t2,t3,t4,t0。对所有测试使用相同的粘度计,对所有测试溶液测量三次。若三个测量值的偏差在平均值的0.35%以内,并且t1是相对于t0的1.6倍至1.8倍,则该值被确定为有效。若不是合适的结果,再次测试缓冲液和样品溶液。
通过如上所述的实验获得的特性粘度值如下表2所示。
如表2所示,分子量越低,发现特性粘度值越低,由此可以看出分子量越小,含水能力越低。
实验例4.使用动物样品的皮肤渗透测试
实施例4中制备的透明质酸钠,lifecore的分子量为200kda透明质酸及比较例7(市售化妆品级透明质酸钠(bloomagefredabiopharm.产品,分子量500,000至120,0000道尔顿)上附着荧光染料:flamma496-二氯三嗪和flamma648-二氯三嗪,对balb/c-裸鼠(雄性,5周龄,14只小鼠)进行皮肤渗透性试验。测试方法如下。
1.测量设备
-ivissepctrum(制造商:perkinelmer,产品编号:124262)
-双光子显微镜(制造商:leicamicrosystems)
2.药物的荧光标记
a.在pbs中以12.5ug/ml的浓度制备实施例4,对照组和比较例7的样品。
b.将荧光染料(flamma496-二氯三嗪,flamma648-二氯三嗪)以1mg/ml的浓度溶解在dmso中。在反应前立即制备并使用新鲜的。
c.将20μl的b荧光染料溶液加入1ml的a的药物溶液中。
d.在室温下培养2小时。每15分钟翻转一次。
3.使用光学成像确认药物皮肤渗透
将实施例4,对照组和比较例7的荧光标记样品以10μl(10μg/ml)施用于balb/c裸鼠背部,小鼠的左侧经过15分钟后,右侧经过30分钟后用pbs缓冲液清洗去除残留物。然后经过30分钟后,1小时后,2小时后,6小时后,24小时后立即用ivissepctrum测量。结果如图2所示。
如图6所示,在对照组和实施例4的情况下,可以看出在30分钟后比15分钟后更有效地渗透皮肤组织。然而,由于比较例7的分子量大于实施例4,因此,不渗透皮肤的情况下被洗掉。
4.通过双光子显微镜(tpm)成像确认药物皮肤渗透
将实施例4,对照组和比较例7的荧光标记样品以10μl(10μg/ml)施用于balb/c裸鼠背部,15分钟后用pbs缓冲液清洗去除残留物。提取小鼠的皮肤切片并用4%甲醛固定制备切口标本,然后使用双光子显微镜对皮肤组织切片进行共焦成像。
如图7所示,当切割皮肤组织并确认使用tpm成像的药物的皮肤渗透时,发现实施例4和对照组相对渗入皮肤组织,然而,比较例7几乎没在皮肤组织内。
如上所述,本领域技术人员将理解,本发明可以以各种修改的形式实现。因此,上述实施例在所有方面都应理解为说明性的而非限制性的。本发明的范围由以下权利要求而不是详细描述示出,并且从权利要求的含义和范围以及等同概念得出的所有改变或修改都包括在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!