四氮唑功能化的UIO-67的合成方法及其应用与流程
本发明涉及多孔吸附材料技术领域,具体涉及一种四氮唑功能化的uio-67的合成方法及其应用。
背景技术:
co2作为温室气体是导致全球变暖的主要原因之一,人类必须面对的主要挑战之一是努力开发更有效的二氧化碳捕获过程。化石燃料导致的co2排放量占据全球co2排放量的80%,随着工业的发展,其排放量可能还进一步提高。因此,控制和减缓因石化燃料使用而向大气中排放的co2是co2减排的重点。工厂的烟道气排放中除含有15%左右的co2外,还含有约75%的n2,能够在混合气体中选择性地吸附co2对于co2的捕捉才是至关重要的。湿法洗涤捕获co2即通过胺溶液(例如单乙醇胺)化学吸收co2,是目前在工业上大规模使用的主要技术。然而,由于形成强烈的化学键(化学吸附),胺对二氧化碳虽然具有高选择性,但是同时也存在着高耗能、高成本等缺点。
利用多孔材料如活性炭和沸石吸附封存co2的方案被认为是一种经济的、可行的co2减排方案。金属有机框架(metalorganicframeworks,mofs)作为一种新型多孔材料,具有高的比表面积和孔隙率,尤其是孔道结构可剪裁易功能化等优点,因此在co2捕集和存储方面受到了越来越多的关注。通过在mofs孔道中引入不饱和金属位点,极性位点、碱性位点等功能位点,人们已制备出一系列具有较高co2/n2吸附选择性的材料,然而大部分mofs材料稳定性不高,在有水汽存在的条件下或长时间暴漏于空气中容易失去结晶性,严重限制了mofs材料在该领域的应用。
zr-mofs是由zr4+和羧酸配体在溶剂热条件下合成出的一类新型mofs,具有很高的热稳定性和化学稳定性,尤其是水稳定性,在酸碱长时间处理后仍能保持很高的结晶性和比表面积,而且合成方法简单,提高了其进一步应用的可能。uio系列zr-mofs如uio-66,uio-67,配体简单易得,合成方法简单,是目前受到广泛关注的mofs材料。其中uio-67配体长度更长,孔道更大,比表面积也更高,更容易在孔道中进行修饰来调节其性能。然而uio-67材料在co2/n2的吸附分离方面存在以下缺点:
1)在低压区,如0.15bar附近对co2的吸附量较低,工厂烟道气中co2的压力大概在0.15bar附近,在该压力下材料对co2的吸附容量值更能评价材料对co2的吸附性能;
2)对co2/n2的吸附选择性较低。
因此研发一种功能化的uio-67材料,一方面提高低压区对co2的吸附容量,另一方面对n2不吸附,从而解决了uio-67材料在该方面应用的缺陷,将对捕集工厂烟气排放中的co2和存储具有重要意义。
技术实现要素:
为解决上述问题,本发明的目的是提供一种四氮唑功能化的uio-67的合成方法,本发明通过在配体上引入氨甲基四氮唑,合成出氨甲基四氮唑功能化的uio-67材料,在对联苯二甲酸配体中引入醛基,利用氨甲基四氮唑与其反应,并进一步还原,合成新型的四氮唑功能化的联苯二甲酸配体,并进一步与zr4+反应,制备出四氮唑功能化的uio-67多孔吸附材料,从而解决了uio-67材料在co2/n2的吸附分离方面的缺点。
为达到上述目的,本发明采用了以下技术方案:
一种四氮唑功能化的uio-67的合成方法,包括如下步骤:
所述四氮唑功能化的uio-67所用配体的合成路线为:
所述四氮唑功能化的uio-67的合成方法包括如下步骤:
(1)配体四氮唑功能化的联苯二甲酸的合成
(a)甲基对联苯二甲酸甲酯(a1)的合成:将甲基对联苯二甲酸加入到盛有甲醇的反应容器中,向反应体系中缓慢滴加浓硫酸,滴加完后,加热回流反应完成后,冷却至室温,旋蒸至出现大量固体析出,过滤,洗涤,干燥,得到白色固体即为甲基对联苯二甲酸甲酯(a1);
(b)二溴甲基联苯二甲酸甲酯(a2)的合成:将甲基对联苯二甲酸甲酯(a1)溶解到四氯化碳溶剂中,室温下通氩气,然后在惰性气氛下依次向反应体系中加入n-溴代丁二酰亚胺和偶氮二异丁腈,加热回流反应完成后,冷却至室温,过滤,滤液分别用饱和亚硫酸钠溶液、饱和碳酸氢钠溶液、饱和氯化钠溶液和水洗涤,分离出有机层,用无水硫酸钠干燥干燥,过滤,滤液旋干,加入甲醇回流,过滤干燥,得到白色固体即为二溴甲基联苯二甲酸甲酯(a2);
(c)醛基联苯二甲酸甲酯(a3)的合成:将二溴甲基联苯二甲酸甲酯(a2)加入丙酮/水混合溶剂中,向反应体系中加入硝酸银,避光,室温下反应,反应结束后将丙酮蒸出,向反应体系中加入二氯甲烷萃取,分离出有机层,有机层用饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,蒸干,得到白色固体二溴甲基联苯二甲酸甲酯(a3);
(d)四氮唑功能化的联苯二甲酸甲酯(a4)的合成:将胺甲基四氮唑溶解在甲醇中,室温下加入醛基联苯二甲酸甲酯(a3),回流反应后,冷却至室温,加入氰基硼氢化钠,加热反应,反应完后向体系中加入饱和碳酸钠溶液,将甲醇蒸出,向反应体系中加入水,用乙醚萃取,有机层用水洗涤,无水硫酸钠干燥,蒸干,继续向反应体系中加入二氯甲烷,搅拌,静置,将沉淀过滤,得到白色固体即为四氮唑功能化的联苯二甲酸甲酯(a4);
(e)四氮唑功能化的联苯二甲酸(a5)的合成:将四氮唑功能化的联苯二甲酸甲酯(a4)溶解在四氢呋喃中,向反应体系中加入1mol/l氢氧化钠溶液,回流反应后,冷却至室温,将四氢呋喃蒸出,用1mol/l的盐酸溶液将体系ph值调至酸性,析出大量白色沉淀,过滤,沉淀用水洗涤,干燥,得到白色固体即为四氮唑功能化的联苯二甲酸(a5);
(2)配合物四氮唑功能化的uio-67的合成
所述配合物四氮唑功能化的uio-67的合成路线为:
所述配合物四氮唑功能化的uio-67的合成方法如下:
将zrcl4加入到盛有n,n-二甲基甲酰胺的反应釜中,加入浓盐酸,超声处理,使zrcl4完全溶解,继续向反应体系中加入n,n-二甲基甲酰胺,然后加入配体四氮唑功能化的联苯二甲酸(a5),再次超声处理,配体完全溶解后,加热反应,反应完成后,过滤,沉淀用n,n-二甲基甲酰胺洗涤至上清液为无色,丙酮洗涤,干燥,即得到四氮唑功能化的uio-67。此反应具有反应条件温和、易于提纯分离的优点。
进一步的,所述步骤(1)第(a)步中,所述甲基对联苯二甲酸与甲醇的用量配比为0.1-0.2mmol:1ml,所述向反应体系中滴加浓硫酸的量与甲基对联苯二甲酸的用量配比为0.07-0.08ml:1mmol,所述加热反应的温度为75-85℃,回流反应时间为11-13h。此反应的反应条件温和,方法简单,易于提纯分离,产率较高。
再进一步的,所述步骤(1)第(b)步中,所述甲基对联苯二甲酸甲酯(a1)与四氯化碳的用量配比为0.1-0.3mmol:1ml,所述室温下通氩气时间为25-35min,所述在惰性气氛下加入的n-溴代丁二酰亚胺的量为甲基对联苯二甲酸甲酯(a1)的1-3倍,加入的偶氮二异丁腈的量为甲基对联苯二甲酸甲酯(a1)的0.6%-0.7%,所述的加热回流反应温度为75-85℃,反应时间为9-11h,所述甲醇回流中甲醇用量与甲基对联苯二甲酸甲酯(a1)的配比3-4ml:1mmol,回流时间为1-3h。此反应反应条件温和,方法简单,易于提纯分离,产率较高。
更进一步的,所述步骤(1)第(c)步中,所述丙酮/水混合溶剂的体积比为4:1,二溴甲基联苯二甲酸甲酯(a2)与丙酮/水混合溶剂的用量配比为1mmol:10ml,所述向反应体系中加入硝酸银的摩尔量为二溴甲基联苯二甲酸甲酯(a2)的1.2-3.2倍,室温下反应时间为24h,所述萃取用二氯甲烷与二溴甲基联苯二甲酸甲酯(a2)的用量配比为10ml:1mmol。此反应反应条件温和,方法简单,易于提纯分离,产率较高。
更进一步的,所述步骤(1)第(d)步中,所述甲醇与胺甲基四氮唑的用量配比为6ml:1mmol,加入的胺甲基四氮唑的摩尔量是醛基联苯二甲酸甲酯(a3)的1-2倍,回流反应时间为4-6h,加入的氰基硼氢化钠的摩尔量为胺甲基四氮唑的1.5-2.5倍,加热反应的温度为45-55℃,加热反应时间为4.5-5.5h,反应完后向体系中加入的饱和碳酸钠溶液与胺甲基四氮唑的配比为1.5-2.5ml:1mmol,加入的水与胺甲基四氮唑的配比为3.5-4.5ml:1mmol,用乙醚萃取次数为3次,每次乙醚用量与胺甲基四氮唑的配比为5.5-6.5ml:1mmol。此反应反应条件温和,方法简单,易于提纯分离,产率较高。
更进一步的,所述步骤(1)第(e)步中,所述四氮唑功能化的联苯二甲酸甲酯(a4)与四氢呋喃的用量配比为0.2-0.4mmol:1ml,1mol/l氢氧化钠溶液与四氮唑功能化的联苯二甲酸甲酯(a4)的用量配比为6-8ml:1mmol,所述回流反应时间为4.5-5.5h,用1mol/l的盐酸调节ph值至2-5。此反应反应条件温和,方法简单,易于提纯分离,产率较高。
更进一步的,所述步骤(2)中,将zrcl4加入到盛有n,n-二甲基甲酰胺的反应釜中,其中zrcl4与反应釜中n,n-二甲基甲酰胺的用量配比为0.07-0.09mmol:1ml,加入的浓盐酸的与zrcl4的用量配比为1.2-1.3ml:1mmol,超声处理时间为18-22min,使zrcl4完全溶解之后,所述继续向反应体系中加入的n,n-二甲基甲酰胺量与zrcl4的用量配比为24.5-25.5ml:1mmol,加入的配体四氮唑功能化的联苯二甲酸(a5)的摩尔量与zrcl4等量,再次超声处理时间为8-12min,加热反应的温度75-85℃,反应时间为23-25h,所述丙酮洗涤次数为3次。此反应反应条件温和,方法简单,易于提纯分离,产率较高。
一种四氮唑功能化的uio-67的应用,其中所述四氮唑功能化的uio-67可应用于混合气体中co2的选择性捕捉和储存。
与现有技术相比,本发明具有以下有益效果:
1、本发明合成的配合物四氮唑功能化的uio-67为纯相并且具有很高的结晶性,孔道规整,有利于气体分子在孔道中的渗透和传递。
2、本发明合成的配合物四氮唑功能化的uio-67材料具有较高的热稳定性和化学稳定性,有利于实际应用条件下延长材料的使用寿命。
3、uio-67对于n2的吸附为阶梯性质,本发明合成的配合物四氮唑功能化的uio-67由于引入的四氮唑功能基团尺寸较大,占据了uio-67的孔道,使孔道的尺寸减小,克服了uio-67阶梯状吸附现象,完全为微孔吸附性质,有利于降低co2/n2混合气中对尺寸较大的n2的吸附。
4、本发明合成的配合物氨甲基四氮唑功能化的uio-67,在孔道中引入四氮唑功能基团后,增大了材料与co2的相互作用力,在co2压力较低时,明显提高了对co2的吸附能力。
5、本发明合成的配合物氨甲基四氮唑功能化的uio-67,在低压力下对co2具有良好的吸附性能,而在整个吸附压力范围内对n2基本不吸附,在实际工作条件下对co2/n2的吸附选择性较强,非常值得推广应用于工厂烟道混合气体中co2的捕捉和储存。
6、本发明的合成方法简单,成本低,易于推广应用。
附图说明
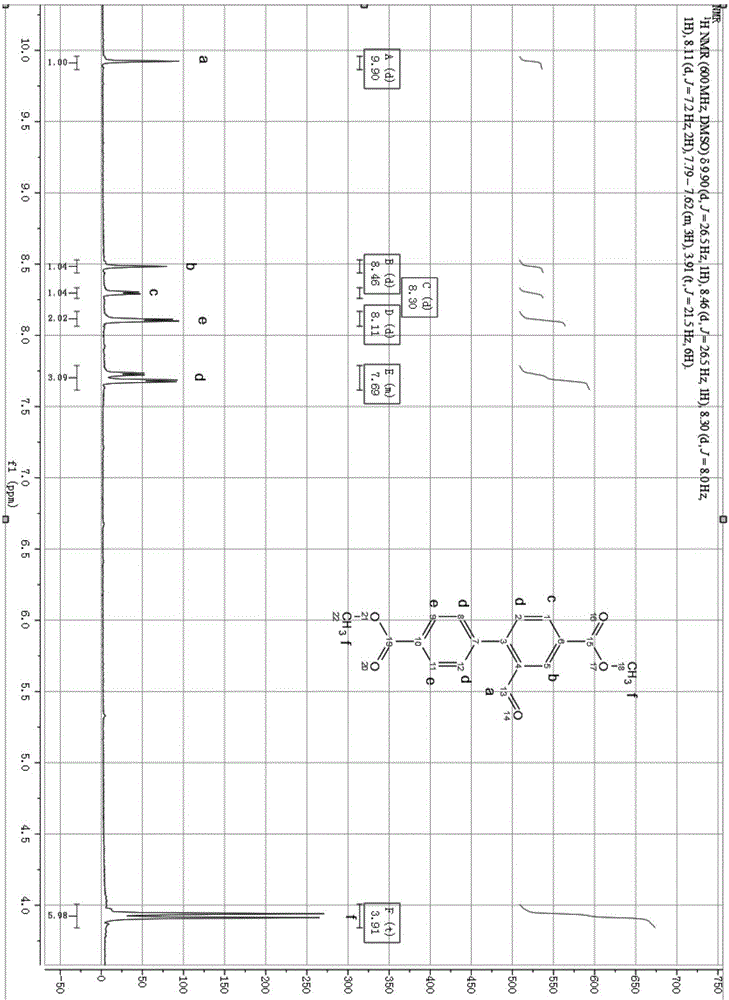
图1为本发明醛基联苯二甲酸甲酯的核磁谱图;
图2为四氮唑功能化的联苯二甲酸的核磁谱;
图3为四氮唑功能化的uio-67(uio-67-tetra)的x射线粉末衍射图;
图4为四氮唑功能化的uio-67(uio-67-tetra)热重分析曲线;
图5为四氮唑功能化的uio-67(uio-67-tetra)和uio-67在77k下n2的吸脱附等温线;
图6为四氮唑功能化的uio-67(uio-67-tetra)和uio-67在273k对co2和n2的吸附曲线;
图7和图8为四氮唑功能化的uio-67(uio-67-tetra)和uio-67在273k低压区对co2和n2的吸附曲线;
具体实施方式
为了进一步阐述本发明的技术方案,下面结合附图及实施例对本发明进行进一步说明。
实施例1
一种四氮唑功能化的uio-67的合成方法,包括如下步骤:
所述四氮唑功能化的uio-67所用配体的合成路线为:
所述四氮唑功能化的uio-67的合成方法包括如下步骤:
(1)配体四氮唑功能化的联苯二甲酸的合成
(a)、甲基对联苯二甲酸甲酯(a1)的合成:称取20mmol甲基对联苯二甲酸加入到盛有150ml甲醇的反应容器中,缓慢滴加1.5ml的浓硫酸,滴加完后,加热至80℃回流反应12h,冷却至室温,旋蒸至出现大量固体析出,过滤,并用冷冻甲醇洗涤,干燥,得到白色固体即为甲基对联苯二甲酸甲酯(a1),产率95%。
(b)、二溴甲基联苯二甲酸甲酯(a2)的合成:称取15mmol的甲基对联苯二甲酸甲酯(a1)溶解到80ml的四氯化碳溶剂中,室温下通氩气30min,然后在惰性气氛下依次加入32mmol的n-溴代丁二酰亚胺nbs和0.1mmol的偶氮二异丁腈aibn,加热至80℃反应10h,冷却至室温,过滤,滤液分别用饱和亚硫酸钠溶液、饱和碳酸氢钠溶液、饱和氯化钠溶液和水洗涤,分离出有机层,用无水硫酸钠干燥,过滤,滤液旋干,然后加入50ml甲醇回流2h,过滤干燥,得到白色固体即为二溴甲基联苯二甲酸甲酯(a2),产率90%。
(c)、醛基联苯二甲酸甲酯(a3)的合成:称取10mmol的二溴甲基联苯二甲酸甲酯(a2)至100ml的丙酮/水(v:v=4:1)混合溶剂中,加入22mmol的硝酸银,避光,室温下反应24h,反应结束后将丙酮蒸出,加入100ml二氯甲烷萃取,分离出有机层,有机层用饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,蒸干,得到白色固体即为醛基联苯二甲酸甲酯(a3),产率65%。
(d)、四氮唑功能化的联苯二甲酸甲酯(a4)的合成:称取7.5mmol的胺甲基四氮唑溶解在45ml的甲醇中,室温下加入5mmol的醛基联苯二甲酸甲酯(a3),回流5h后,冷却至室温,加入15mmol的氰基硼氢化钠,在50℃继续反应5h,反应完后向体系中加入15ml的饱和碳酸钠溶液,将甲醇蒸出,加入30ml的水,用乙醚萃取(45ml*3),有机层用水洗涤,无水硫酸钠干燥,蒸干,继续加入二氯甲烷,搅拌,静置,将沉淀过滤,得到白色固体即为四氮唑功能化的联苯二甲酸甲酯(a4),产率70%。
(e)、四氮唑功能化的联苯二甲酸(a5)的合成:称取3mmol四氮唑功能化的联苯二甲酸甲酯(a4)溶解在10ml的四氢呋喃中,加入20ml1m氢氧化钠溶液,回流,反应5h后,冷却至室温,将四氢呋喃蒸出,用1m的盐酸溶液将ph值调至3-4之间,析出大量白色沉淀,过滤,沉淀用水洗涤两次,干燥,得到白色固体a5,产率95%。
(2)、配合物四氮唑功能化的uio-67的合成(uio-67-tetra)
所述配合物四氮唑功能化的uio-67的合成路线为:
称取0.4mmol的zrcl4加入到装有5mln,n-二甲基甲酰胺(dmf)的反应釜中,加入0.5ml的浓盐酸,超声20min,使zrcl4完全溶解,加入10ml的n,n-二甲基甲酰胺(dmf),然后加入0.4mmol的配体四氮唑功能化的联苯二甲酸a5,超声10min,配体完全溶解后,在80℃条件下反应24h,反应完成后,过滤,沉淀用n,n-二甲基甲酰胺(dmf)洗涤至上清液为无色,丙酮洗涤3次,干燥。
实施例2
实施例2与实施例1中所述四氮唑功能化的uio-67所用配体的合成路线相同,所述配合物四氮唑功能化的uio-67的合成路线相同,区别在于:
所述四氮唑功能化的uio-67的合成方法包括如下步骤:
(1)配体四氮唑功能化的联苯二甲酸的合成
(a)、甲基对联苯二甲酸甲酯(a1)的合成:称取15mmol甲基对联苯二甲酸加入到盛有150ml甲醇的反应容器中,缓慢滴加1.05ml的浓硫酸,滴加完后,加热至75℃回流反应11h,冷却至室温,旋蒸至出现大量固体析出,过滤,并用冷冻甲醇洗涤,干燥,得到白色固体即为甲基对联苯二甲酸甲酯(a1),产率90%。
(b)、二溴甲基联苯二甲酸甲酯(a2)的合成:称取8mmol的甲基对联苯二甲酸甲酯(a1)溶解到80ml的四氯化碳溶剂中,室温下通氩气25min,然后在惰性气氛下依次加入8mmol的n-溴代丁二酰亚胺nbs和0.05mmol的偶氮二异丁腈aibn,加热至75℃反应9h,冷却至室温,过滤,滤液分别用饱和亚硫酸钠溶液、饱和碳酸氢钠溶液、饱和氯化钠溶液和水洗涤,分离出有机层,用无水硫酸钠干燥,过滤,滤液旋干,然后加入24ml甲醇回流1.0h,过滤干燥,得到白色固体即为二溴甲基联苯二甲酸甲酯(a2),产率88%。
(c)、醛基联苯二甲酸甲酯(a3)的合成:称取10mmol的二溴甲基联苯二甲酸甲酯(a2)至100ml的丙酮/水(v:v=4:1)混合溶剂中,加入12mmol的硝酸银,避光,室温下反应24h,反应结束后将丙酮蒸出,加入100ml二氯甲烷萃取,分离出有机层,有机层用饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,蒸干,得到白色固体即为醛基联苯二甲酸甲酯(a3),产率63%。
(d)、四氮唑功能化的联苯二甲酸甲酯(a4)的合成:称取5mmol的胺甲基四氮唑溶解在30ml的甲醇中,室温下加入5mmol的醛基联苯二甲酸甲酯(a3),回流4h后,冷却至室温,加入7.5mmol的氰基硼氢化钠,在45℃继续反应4h,反应完后向体系中加入7.5ml的饱和碳酸钠溶液,将甲醇蒸出,加入17.5ml的水,用乙醚萃取(27.5ml*3),有机层用水洗涤,无水硫酸钠干燥,蒸干,继续加入二氯甲烷,搅拌,静置,将沉淀过滤,得到白色固体即为四氮唑功能化的联苯二甲酸甲酯(a4),产率68%。
(e)、四氮唑功能化的联苯二甲酸(a5)的合成:称取2mmol四氮唑功能化的联苯二甲酸甲酯(a4)溶解在10ml的四氢呋喃中,加入12ml1m氢氧化钠溶液,回流,反应4.5h后,冷却至室温,将四氢呋喃蒸出,用1m的盐酸溶液将ph值调至2-3之间,析出大量白色沉淀,过滤,沉淀用水洗涤两次,干燥,得到白色固体a5,产率95%。
(2)、配合物四氮唑功能化的uio-67的合成(uio-67-tetra)
称取0.35mmol的zrcl4加入到装有5mln,n-二甲基甲酰胺(dmf)的反应釜中,加入0.42ml的浓盐酸,超声18min,使zrcl4完全溶解,加入8.6ml的n,n-二甲基甲酰胺(dmf),然后加入0.35mmol的配体四氮唑功能化的联苯二甲酸a5,超声8min,配体完全溶解后,在75℃条件下反应23h,反应完成后,过滤,沉淀用n,n-二甲基甲酰胺(dmf)洗涤至上清液为无色,丙酮洗涤3次,干燥。
实施例3
实施例3与实施例1中所述四氮唑功能化的uio-67所用配体的合成路线相同,所述配合物四氮唑功能化的uio-67的合成路线相同,区别在于:
所述四氮唑功能化的uio-67的合成方法包括如下步骤:
(1)配体四氮唑功能化的联苯二甲酸的合成
(a)、甲基对联苯二甲酸甲酯(a1)的合成:称取30mmol甲基对联苯二甲酸加入到盛有150ml甲醇的反应容器中,缓慢滴加2.4ml的浓硫酸,滴加完后,加热至85℃回流反应13h,冷却至室温,旋蒸至出现大量固体析出,过滤,并用冷冻甲醇洗涤,干燥,得到白色固体即为甲基对联苯二甲酸甲酯(a1),产率92%。
(b)、二溴甲基联苯二甲酸甲酯(a2)的合成:称取24mmol的甲基对联苯二甲酸甲酯(a1)溶解到80ml的四氯化碳溶剂中,室温下通氩气35min,然后在惰性气氛下依次加入72mmol的n-溴代丁二酰亚胺nbs和0.17mmol的偶氮二异丁腈aibn,加热至85℃反应11h,冷却至室温,过滤,滤液分别用饱和亚硫酸钠溶液、饱和碳酸氢钠溶液、饱和氯化钠溶液和水洗涤,分离出有机层,用无水硫酸钠干燥,过滤,滤液旋干,然后加入96ml甲醇回流3h,过滤干燥,得到白色固体即为二溴甲基联苯二甲酸甲酯(a2),产率85%。
(c)、醛基联苯二甲酸甲酯(a3)的合成:称取10mmol的二溴甲基联苯二甲酸甲酯(a2)至100ml的丙酮/水(v:v=4:1)混合溶剂中,加入32mmol的硝酸银,避光,室温下反应24h,反应结束后将丙酮蒸出,加入100ml二氯甲烷萃取,分离出有机层,有机层用饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,蒸干,得到白色固体即为醛基联苯二甲酸甲酯(a3),产率68%。
(d)、四氮唑功能化的联苯二甲酸甲酯(a4)的合成:称取10mmol的胺甲基四氮唑溶解在60ml的甲醇中,室温下加入5mmol的醛基联苯二甲酸甲酯(a3),回流6h后,冷却至室温,加入25mmol的氰基硼氢化钠,在55℃继续反应6h,反应完后向体系中加入25ml的饱和碳酸钠溶液,将甲醇蒸出,加入45ml的水,用乙醚萃取(65ml*3),有机层用水洗涤,无水硫酸钠干燥,蒸干,继续加入二氯甲烷,搅拌,静置,将沉淀过滤,得到白色固体即为四氮唑功能化的联苯二甲酸甲酯(a4),产率70%。
(e)、四氮唑功能化的联苯二甲酸(a5)的合成:称取4mmol四氮唑功能化的联苯二甲酸甲酯(a4)溶解在10ml的四氢呋喃中,加入32ml1m氢氧化钠溶液,回流,反应5.5h后,冷却至室温,将四氢呋喃蒸出,用1m的盐酸溶液将ph值调至4-5之间,析出大量白色沉淀,过滤,沉淀用水洗涤两次,干燥,得到白色固体a5,产率90%。
(2)、配合物四氮唑功能化的uio-67的合成(uio-67-tetra)
称取0.45mmol的zrcl4加入到装有5mln,n-二甲基甲酰胺(dmf)的反应釜中,加入0.60ml的浓盐酸,超声22min,使zrcl4完全溶解,加入11.5ml的n,n-二甲基甲酰胺(dmf),然后加入0.45mmol的配体四氮唑功能化的联苯二甲酸a5,超声12min,配体完全溶解后,在85℃条件下反应25h,反应完成后,过滤,沉淀用n,n-二甲基甲酰胺(dmf)洗涤至上清液为无色,丙酮洗涤3次,干燥。
实施例4
实施例4与实施例1中所述四氮唑功能化的uio-67所用配体的合成路线相同,所述配合物四氮唑功能化的uio-67的合成路线相同,区别在于:
所述四氮唑功能化的uio-67的合成方法包括如下步骤:
(1)配体四氮唑功能化的联苯二甲酸的合成
(a)甲基对联苯二甲酸甲酯(a1)的合成:称取17.3mmol甲基对联苯二甲酸加入到盛有150ml甲醇的反应容器中,缓慢滴加1.3ml的浓硫酸,滴加完后,加热至77℃回流反应13h,冷却至室温,旋蒸至出现大量固体析出,过滤,并用冷冻甲醇洗涤,干燥,得到白色固体即为甲基对联苯二甲酸甲酯(a1),产率85%。
(b)二溴甲基联苯二甲酸甲酯(a2)的合成:称取12mmol的甲基对联苯二甲酸甲酯(a1)溶解到80ml的四氯化碳溶剂中,室温下通氩气27min,然后在惰性气氛下依次加入18mmol的n-溴代丁二酰亚胺nbs和0.08mmol的偶氮二异丁腈aibn,加热至77℃反应9.5h,冷却至室温,过滤,滤液分别用饱和亚硫酸钠溶液、饱和碳酸氢钠溶液、饱和氯化钠溶液和水洗涤,分离出有机层,用无水硫酸钠干燥,过滤,滤液旋干,然后加入38ml甲醇回流1.5h,过滤干燥,得到白色固体即为二溴甲基联苯二甲酸甲酯(a2),产率85%。
(c)醛基联苯二甲酸甲酯(a3)的合成:称取10mmol的二溴甲基联苯二甲酸甲酯(a2)至100ml的丙酮/水(v:v=4:1)混合溶剂中,加入17mmol的硝酸银,避光,室温下反应24h,反应结束后将丙酮蒸出,加入100ml二氯甲烷萃取,分离出有机层,有机层用饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,蒸干,得到白色固体即为醛基联苯二甲酸甲酯(a3),产率68%。
(d)四氮唑功能化的联苯二甲酸甲酯(a4)的合成:称取6mmol的胺甲基四氮唑溶解在36ml的甲醇中,室温下加入5mmol的醛基联苯二甲酸甲酯(a3),回流4.5h后,冷却至室温,加入11mmol的氰基硼氢化钠,在47℃继续反应4.5h,反应完后向体系中加入11ml的饱和碳酸钠溶液,将甲醇蒸出,加入25ml的水,用乙醚萃取(36ml*3),有机层用水洗涤,无水硫酸钠干燥,蒸干,继续加入二氯甲烷,搅拌,静置,将沉淀过滤,得到白色固体即为四氮唑功能化的联苯二甲酸甲酯(a4),产率72%。
(e)四氮唑功能化的联苯二甲酸(a5)的合成:称取2.5mmol四氮唑功能化的联苯二甲酸甲酯(a4)溶解在10ml的四氢呋喃中,加入15.8ml1m氢氧化钠溶液,回流,反应4.7h后,冷却至室温,将四氢呋喃蒸出,用1m的盐酸溶液将ph值调至3-3.5之间,析出大量白色沉淀,过滤,沉淀用水洗涤两次,干燥,得到白色固体a5,产率89%。
(2)配合物四氮唑功能化的uio-67的合成(uio-67-tetra)
称取0.38mmol的zrcl4加入到装有5mln,n-二甲基甲酰胺(dmf)的反应釜中,加入0.46ml的浓盐酸,超声19min,使zrcl4完全溶解,加入9.4ml的n,n-二甲基甲酰胺(dmf),然后加入0.38mmol的配体四氮唑功能化的联苯二甲酸a5,超声9min,配体完全溶解后,在77℃条件下反应23.5h,反应完成后,过滤,沉淀用n,n-二甲基甲酰胺(dmf)洗涤至上清液为无色,丙酮洗涤3次,干燥。
实施例5
实施例5与实施例1中所述四氮唑功能化的uio-67所用配体的合成路线相同,所述配合物四氮唑功能化的uio-67的合成路线相同,区别在于:
所述四氮唑功能化的uio-67的合成方法包括如下步骤:
(1)配体四氮唑功能化的联苯二甲酸的合成
(a)甲基对联苯二甲酸甲酯(a1)的合成:称取26mmol甲基对联苯二甲酸加入到盛有150ml甲醇的反应容器中,缓慢滴加2.0ml的浓硫酸,滴加完后,加热至83℃回流反应12.5h,冷却至室温,旋蒸至出现大量固体析出,过滤,并用冷冻甲醇洗涤,干燥,得到白色固体即为甲基对联苯二甲酸甲酯(a1),产率92%。
(b)二溴甲基联苯二甲酸甲酯(a2)的合成:称取20mmol的甲基对联苯二甲酸甲酯(a1)溶解到80ml的四氯化碳溶剂中,室温下通氩气32min,然后在惰性气氛下依次加入50mmol的n-溴代丁二酰亚胺nbs和0.14mmol的偶氮二异丁腈aibn,加热至83℃反应10.5h,冷却至室温,过滤,滤液分别用饱和亚硫酸钠溶液、饱和碳酸氢钠溶液、饱和氯化钠溶液和水洗涤,分离出有机层,用无水硫酸钠干燥,过滤,滤液旋干,然后加入75ml甲醇回流2.5h,过滤干燥,得到白色固体即为二溴甲基联苯二甲酸甲酯(a2),产率87%。
(c)醛基联苯二甲酸甲酯(a3)的合成:称取10mmol的二溴甲基联苯二甲酸甲酯(a2)至100ml的丙酮/水(v:v=4:1)混合溶剂中,加入27mmol的硝酸银,避光,室温下反应24h,反应结束后将丙酮蒸出,加入100ml二氯甲烷萃取,分离出有机层,有机层用饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,蒸干,得到白色固体即为醛基联苯二甲酸甲酯(a3),产率60%。
(d)四氮唑功能化的联苯二甲酸甲酯(a4)的合成:称取8.7mmol的胺甲基四氮唑溶解在52ml的甲醇中,室温下加入5mmol的醛基联苯二甲酸甲酯(a3),回流5.5h后,冷却至室温,加入20mmol的氰基硼氢化钠,在52℃继续反应5.5h,反应完后向体系中加入20ml的饱和碳酸钠溶液,将甲醇蒸出,加入37ml的水,用乙醚萃取(55ml*3),有机层用水洗涤,无水硫酸钠干燥,蒸干,继续加入二氯甲烷,搅拌,静置,将沉淀过滤,得到白色固体即为四氮唑功能化的联苯二甲酸甲酯(a4),产率75%。
(e)四氮唑功能化的联苯二甲酸(a5)的合成:称取3.5mmol四氮唑功能化的联苯二甲酸甲酯(a4)溶解在10ml的四氢呋喃中,加入26ml1m氢氧化钠溶液,回流,反应5.2h后,冷却至室温,将四氢呋喃蒸出,用1m的盐酸溶液将ph值调至3.5-4之间,析出大量白色沉淀,过滤,沉淀用水洗涤两次,干燥,得到白色固体a5,产率91%。
(2)配合物四氮唑功能化的uio-67的合成(uio-67-tetra)
称取0.43mmol的zrcl4加入到装有5mln,n-二甲基甲酰胺(dmf)的反应釜中,加入0.54ml的浓盐酸,超声21min,使zrcl4完全溶解,加入10.7ml的n,n-二甲基甲酰胺(dmf),然后加入0.43mmol的配体四氮唑功能化的联苯二甲酸a5,超声11min,配体完全溶解后,在83℃条件下反应24.5h,反应完成后,过滤,沉淀用n,n-二甲基甲酰胺(dmf)洗涤至上清液为无色,丙酮洗涤3次,干燥。
上述实施例通过在对联苯二甲酸配体上引入四氮唑功能基团,成功的合成出四氮唑功能化的uio-67-tetra材料,273k下的气体吸附表征表明,四氮唑功能化能够明显提高uio-67材料在低压下对co2的吸附量,而且大大提高co2/n2的吸附选择性,从而证明四氮唑功能化是一种成功的提高co2/n2吸附选择性的策略,考虑到zr-mofs材料的稳定性,四氮唑功能化的uio-67可应用于燃烧后co2的捕捉和储存。
合成方法验证与合成物的性能及应用分析如下:
1、四氮唑功能化的联苯二甲酸的合成
附图1是醛基联苯二甲酸甲酯(a3)核磁谱,醛基联苯二甲酸甲酯(a3):1hnmr(600hz,d-dmso)δ9.90(d,j=26.5hz,1h),8.46(d,j=26.5hz,1h),8.30(d,j=8.0hz,1h),8.11(d,j=7.2hz,2h),7.79-7.62(m,3h),3.91(t,j=21.5hz,6h)。
附图2是四氮唑功能化的联苯二甲酸(a5)的核磁谱,四氮唑功能化的联苯二甲酸(a5):1hnmr(600hz,d-dmso)δ8.32(s,1h),7.98(t,j=10.5hz,2h),7.95(d,j=7.8hz,1h),7.52(d,j=8.0hz,2h),7.42(d,j=7.9hz,1h),4.07(s,2h),3.88(s,2h)。
核磁结果表明,本实施例已成功合成出四氮唑功能化的联苯二甲酸配体。
2、配合物四氮唑功能化的uio-67(uio-67-tetra)的合成
金属盐四氯化锆和配体四氮唑功能化的联苯二甲酸通过溶剂热一锅法直接合成配合物四氮唑功能化的uio-67(uio-67-tetra)。
3、x射线粉末衍射分析
对配合物四氮唑功能化的uio-67样品(uio-67-tetra)进行x射线粉末衍射测试,结果如图3所示。其x射线粉末衍射图说明该样品为纯相并且具有很高的结晶性。与uio-67相比没有发生明显的变化,表明在配体上引入较大的功能基团四氮唑后,并不影响配体中的羧基与zr4+的配位模式与构型,仍然可以合成出与uio-67同构的新型框架结构。
4、热稳定性分析
在氮气气氛条件下,对配合物四氮唑功能化的uio-67(uio-67-tetra)样品做热重分析测试,具体结果如附图4所示。从附图4中可以看出,四氮唑功能化的uio-67显示出较高的热稳定性。
5、配合物四氮唑功能化的uio-67(uio-67-tetra)的吸附分析
在77k条件下对配合物四氮唑功能化的uio-67(uio-67-tetra)的n2吸附性质进行分析,在分析前首先将样品在200℃下脱气10小时。
uio-67-tetra的n2吸附如附图5所示。uio-67对于n2的吸附呈现出明显的阶梯性质。功能化后的四氮唑功能化的uio-67(uio-67-tetra),由于引入的四氮唑功能基团尺寸较大,占据了uio-67的孔道,一方面使孔道的尺寸减小,表现在吸附曲线上是阶梯状吸附现象消失,完全表现为微孔吸附的曲线图像。
如附图6至8所示,在273k条件下,尽管在1bar条件下,uio-67对于co2的吸附容量高于氨甲基四氮唑功能化的uio-67(uio-67-tetra),但在低压区尤其是在0.15bar左右的压力下,四氮唑功能化的uio-67(uio-67-tetra)对co2的吸附量明显大于功能化前的uio-67,而在整个压力范围内对n2没有明显吸附。工厂排放的烟道气中,co2的比例约为15%,其余主要为n2,燃烧后co2捕捉和储存一方面要求材料要具有高的co2/n2吸附选择性,另一方面也要求材料在低压区如0.15bar左右对co2有较高的吸附容量。从附图7的低压吸附图上可以看出,在材料孔道中引入四氮唑功能基团后,吸附曲线的斜率增大,表明材料与co2的相互作用较强,而且明显提高了材料在低压区对co2的吸附能力;此外功能化后,材料在整个吸附压力范围内对n2没有明显吸附,因此四氮唑功能化不但能够明显提高uio-67材料在实际工作条件下对co2的吸附容量,而且能明显提高其对co2/n2的吸附选择性。适用于工厂排放的烟道气中co2的捕捉和储存。
- 还没有人留言评论。精彩留言会获得点赞!