一种盐酸甲氯芬酯的制备方法与流程
本发明属于化学合成领域,具体的,本发明涉及一种中枢神经兴奋药盐酸甲氯芬酯的制备方法。
背景技术:
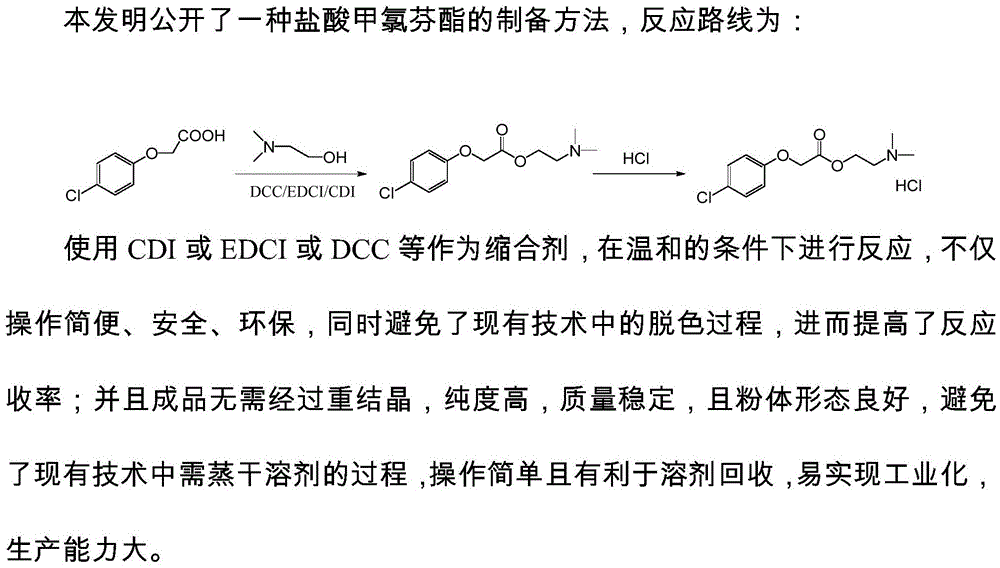
:盐酸甲氯芬酯(meclofenoxatehydrochloride),cas:3685-84-5,商品名为氯酯醒,化学名为对氯苯氧乙酸二甲氨基乙酯盐酸盐(2-(dimethylamino)ethyl4-chlorophenoxyacetatehydrochoride),化学结构式如下所示:盐酸甲氯芬酯主要作用于大脑皮质,它能促进脑细胞的氧化还原,调节神经细胞的代谢,增加对糖类的利用对受抑制的中枢神经有兴奋作用。治疗外伤性昏迷、儿童遗尿症、意识障碍、老年性精神病、各种痴呆、酒精中毒等。目前国内采用的生产工艺有以下几种:中国专利cn201210409046.x于2013.01.16公开了一种盐酸甲氯芬酯的制备方法,其特征在于,使用对氯苯氧乙酸以甲苯为溶剂,在高温下和二氯亚砜、有机碱反应形成季铵盐,后在高温下滴加二甲氨基乙醇,反应完毕后蒸干溶剂得到盐酸甲氯芬酯粗品,粗品在一元醇中重结晶得到盐酸甲氯芬酯成品,路线如下:该方法需要使用苯做溶剂、毒性大、在高温中缓慢滴加二甲氨基乙醇,需要高温反应,且反应宜慢,滴加速度过快会产生副反应,粗品颜色偏黄,且后处理需蒸干溶剂后进行重结晶,工艺相对复杂困难。另外,中国专利cn201410310852.0于2014.10.01公开一种盐酸甲氯芬酯粗品的合成工艺,保护了的是车间生产盐酸甲氯芬酯的合成工艺,具体步骤包括:加热回流,冷却,回流脱色,抽滤和静置分层,该方案需要以甲苯为溶剂高温回流长时间,然后低温静置析晶,路线如下:该方法以甲苯为溶剂高温回流长时间,然后低温静置析晶,操作繁琐。赵普等发表了《盐酸甲氯芬酯的合成》(药学研究,2012,31(12):690-691)。该文献公开了一种以对氯苯氧乙酸和n,n-二甲基乙胺为起始原料,以对甲苯磺酸为催化剂,在甲苯条件下共沸分水,进行酯化反应,制得甲氯芬酯。然后再异丙醇中直接加入二氯亚砜进行成盐,最终制得盐酸甲氯芬酯。该方法使用了高腐蚀性强酸甲苯磺酸,且需要在甲苯条件下共沸分水,后需低温析晶,不易于操作,二氯亚砜过量时污染较大,后续存在成盐酸盐不充分的问题,且收率只有70%,不适用于工业化。综上可知,现有技术制备盐酸甲氯芬酯,多采用高毒高腐蚀试剂,反应条件相对苛刻,操作步骤繁琐,产品纯度及收率均不够理想。因此,提供一种优化的适于大规模生产的盐酸甲氯芬酯制备方法是现有技术没有解决的技术问题,该制备方法避免苯、甲苯等毒性溶剂的使用,反应条件温和,反应步骤简单,避免现有技术中的脱色过程,所得产品的外观颜色符合要求,反应收率高。通过使用乙酸乙酯作为成盐溶剂,提高了产品质量和产品收率,避免了现有技术中需蒸干溶剂的过程,操作简单且有利于溶剂回收。技术实现要素:本发明的目的在于克服现有技术的不足,提供一种盐酸甲氯芬酯的制备方法,该方法在能够获得高收率,高纯度产品的同时,达到操作简单的效果。具体的,本发明所述盐酸甲氯芬酯的制备方法,避免了在高温下使用如甲苯磺酸等强酸以及甲苯等有毒溶剂,使用cdi或edci或dcc等作为缩合剂((cdi(n,n'-carbonyldiimidazole)n,n'-羰基二咪唑、edci(1-(3-dimethylaminopropyl)-3-ethylcarbodiimidehydrochloride)1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、(dcc(dicyclohexylcarbodiimide)二环己基碳二亚胺),在温和的条件下进行反应,不仅操作简便、安全、环保,同时避免了现有技术中的脱色过程,所得产品的外观颜色符合要求,进而提高了反应收率;通过使用乙酸乙酯作为成盐溶剂,提高了产品质量和产品收率,避免了现有技术中需蒸干溶剂的过程,操作简单且有利于溶剂回收。本发明的上述有益效果通过如下方法实现:本发明的盐酸甲氯芬酯的制备方法,其反应路线如下:按如下方法制备:(a)在单口瓶中将对氯苯氧乙酸投入5~20:1=v/w有机溶剂中,后加入二甲氨基乙醇,室温搅拌10~30min,最后加入缩合剂,其中:对氯苯氧乙酸、二甲氨基乙醇与缩合剂比为1:0.8~1.2:0.9~1.3,缩合剂的浓度为0.2~1mol/l,25℃下搅拌反应1~5h;(b)反应结束后加入水淬灭,萃取分层,有机层通过氯化氢气体成盐酸盐,至固体大量析出,在10~30℃下缓慢搅拌1~3h析晶,过滤,烘干,得到白色固体,即为盐酸甲氯芬酯成品。本发明中缩合剂种类配合溶剂及反应体系底物浓度是实现技术效果的关键技术之一,当缩合剂为dcc、edci或者cdi,同时有机溶剂为二氯甲烷或乙酸乙酯,且控制有机溶剂用量为反应体系底物质量的5~20倍时,有利于实现反应的充分进行。具体的,在步骤(a)中,所述有机溶剂为二氯甲烷或乙酸乙酯,对氯苯氧乙酸与有机溶剂的当量比为8~15:1=v/w,室温搅拌时间为20min,对氯苯氧乙酸、二甲氨基乙醇与缩合剂比为1:0.9~1.2:1.0~1.2,缩合剂的浓度为0.3~0.9mol/l,25℃下搅拌2~4h。所述缩合剂为dcc、edci或者cdi。更具体的,在本发明的盐酸甲氯芬酯的制备方法中,在步骤(a)中,所述有机溶剂为乙酸乙酯,对氯苯氧乙酸与有机溶剂的当量比为10:1=v/w,室温搅拌时间为20min,对氯苯氧乙酸、二甲氨基乙醇与缩合剂比为1:1.1:1.1,缩合剂的浓度为0.6mol/l,25℃下搅拌3h。所述缩合剂为edci。本发明中氯化氢气体的流速以及淬灭水的用量也是关键技术之一,两者构成了成盐时氯化氢气体的浓度,氯化氢浓度低,成盐反应速度慢,成盐不充分,氯化氢浓度高,产品颜色偏黄。具体的,在步骤(b)中,反应后加入2~5:1=v/w的水淬灭,萃取分层,有机层通过氯化氢气体成盐酸盐,氯化氢气体的流速为10~18m/s,在25℃下缓慢搅拌2h析晶。更具体的,在步骤(b)中,反应后加入4:1=v/w的水淬灭,氯化氢气体的流速为12~15m/s。较好的是,本发明的盐酸甲氯芬酯的制备方法,包括如下步骤:(a)在单口瓶中将对氯苯氧乙酸投入10:1=v/w乙酸乙酯中,后加入二甲氨基乙醇,室温搅拌20min,最后加入缩合剂edci,对氯苯氧乙酸、二甲氨基乙醇与缩合剂比为1:1.1:1.1,缩合剂的浓度为0.6mol/l,25℃下搅拌反应3h;(b)反应结束后加入4:1=v/w的水淬灭,萃取分层,有机层通过氯化氢气体成盐酸盐,氯化氢气体的流速为15m/s,至固体大量析出,在25℃下缓慢搅拌2h析晶,过滤,烘干,得到白色固体为盐酸甲氯芬酯成品,该粉体疏松,粘性弱,不易粘附反应设备和搅拌桨,便于贮存和运输,且利于工业化放大生产。本发明与现有技术相比具有如下突出的优点及有益效果:1.避免了在高温下使用如甲苯磺酸等强酸以及甲苯等有毒溶剂,使用cdi或edci或dcc等作为缩合剂,在温和的条件下进行反应,不仅操作简便、安全、环保,同时避免了现有技术中的脱色过程,进而提高了反应收率。2.成品无需经过重结晶,纯度高,质量稳定,且粉体形态良好,避免了现有技术中需蒸干溶剂的过程,操作简单且有利于溶剂回收,易实现工业化,生产能力大。具体实施方式:下面结合实施例对本发明作进一步详细的描述,但发明的实施方式不限于此。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。实施例1(a)在单口瓶中将对氯苯氧乙酸(50g,0.26mol)投入500ml乙酸乙酯中,后加入二甲氨基乙醇(25.4g,0.29mol),室温搅拌20min,最后加入缩合剂edci(55.4g,0.29mol),25℃下搅拌反应3h;(b)反应结束后加入200ml水淬灭,萃取分层,有机层通过氯化氢气体成盐酸盐,氯化氢气体的流速为15m/s,至固体大量析出,在25℃下缓慢搅拌2h析晶,过滤,烘干得到白色固体67g,收率87.6%,纯度99.7%,堆密度为0.51g/cm3,休止角为28°,粉体疏松,粘性弱。实施例2(a)在单口瓶中将对氯苯氧乙酸(50g,0.26mol)投入250ml二氯甲烷中,后加入二甲氨基乙醇(20.1g,0.23mol),室温搅拌10min,最后加入缩合剂dcc(47.3g,0.23mol),25℃下搅拌反应1h;(b)反应结束后加入100ml水淬灭,萃取分层,有机层通过氯化氢气体成盐酸盐,氯化氢气体的流速为10m/s,至固体大量析出,在10℃下缓慢搅拌1h析晶,过滤,烘干得到白色固体64g,收率83.7%,纯度99.7%,堆密度为0.55g/cm3,休止角为30°,粉体疏松,粘性弱。实施例3(a)在单口瓶中将对氯苯氧乙酸(50g,0.26mol)投入1000ml乙酸乙酯中,后加入二甲氨基乙醇(27.2g,0.31mol),室温搅拌30min,最后加入缩合剂cdi(55.0g,0.34mol),25℃下搅拌反应5h;(b)反应结束后加入250ml水淬灭,萃取分层,有机层通过氯化氢气体成盐酸盐,氯化氢气体的流速为18m/s,至固体大量析出,在30℃下缓慢搅拌3h析晶,过滤,烘干得到白色固体66g,收率86.3%,纯度99.8%,堆密度为0.58/cm3,休止角为32°,粉体疏松,粘性弱。对比例1(参照cn201210409046.x进行实验)将对氯苯氧乙酸(50g,0.26mol)和n,n-二异丙基乙胺(6.5g,0.05mol)投入到150ml甲苯中,室温搅拌下加入二氯亚砜(34.5g,0.29mol),随后升温至75℃保温30min,继续升温至100℃,温度保持在70~100℃之间,加入二甲氨基乙醇(25.8g,0.27mol),滴加完毕后保温2h,回收溶剂之干,加入无水乙醇加热溶解,加适量活性炭脱色,过滤,冷却搅拌,烘干得到白色结晶体60.2g,收率78.8%,纯度97.4%,堆密度为0.70g/cm3,休止角为38°,粉体部分成块,粘性弱。对比例2(参照cn201410310852.0进行实验)将对氯苯氧乙酸(50g,0.26mol)投入到150ml甲苯中,加入二甲氨基乙醇(20.5g,0.23mol)和对甲苯磺酸(1.5g),搅拌升温至100℃,回流反应8h,反应结束后,温度降至90℃,加入活性炭,回流脱色30min,过滤,滤液降温至5℃后,通入氯化氢气体成盐酸盐,0~4℃下结晶,保温析晶6h,抽滤,烘干,得到白色晶体56.5g,收率74.0%,纯度95.2%,堆密度为0.75g/cm3,休止角为43°,粉体部分成块,略带粘性。对比例3(参照(赵普.盐酸甲氯芬酯的合成[j].药学研究,2012,31(12):690-691.)进行实验)将对氯苯氧乙酸(50g,0.26mol)加入到150ml甲苯中,加入对甲苯磺酸(2.3g),搅拌下滴加二甲氨基乙醇(34.7g,0.39mol),搅拌混合,有固体出现,油浴升温至80℃左右,固体逐渐溶解,继续升温至回流,体系澄清,使用分水器分水,回流反应10h,缓慢降至室温,分别用饱和的nahco3水溶液和食盐水洗涤有机相,静置分液,有机相减压浓缩,得到浅黄色油状物后,加入异丙醇,冰水浴降温至0~5℃,搅拌下缓慢滴加二氯亚砜(26.0g,0.22mol),逐渐析出淡黄色固体,滴加完毕后,继续在0~5℃下搅拌3h,过滤,用0~5℃的100ml异丙醇打浆洗涤滤饼,过滤,烘干,得到淡黄色粉末状固体53.5g,收率70%,纯度96.5%,堆密度为0.77g/cm3,休止角为40°,粉体疏松,略带粘性。表1不同方法制备盐酸甲氯芬酯的收率、纯度、成品休止角以及堆密度比较收率纯度休止角堆密度实施例187.6%99.7%28°0.51g/cm3实施例283.7%99.7%30°0.55g/cm3实施例386.3%99.8%32°0.58g/cm3对比例178.8%97.4%38°0.70g/cm3对比例274.0%95.2%43°0.75g/cm3对比例370.0%96.5%40°0.77g/cm3由表1数据可知,采用本方案的方法制备的盐酸甲氯芬酯,具有收率高,纯度高,且成品颜色佳、形态良好的优点,从测量休止角和堆密度可知,本方案得到的盐酸甲氯芬酯疏松且流动性较好,原因在于本方案采用缩合剂常温反应,避免了高温下使用甲苯等溶剂,反应温和,同时无需脱色以及重结晶过程,因此在产品的收率纯度以及形态上优于对比例的方案,在工业生产上,避免了现有技术中需蒸干溶剂的过程,操作简单且有利于溶剂回收,体现为简便、安全、环保的特点,粉体不易粘附反应设备和搅拌桨,便于贮存和运输,利于后续制剂工业化放大生产。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1