一种光修复性偶氮苯聚合物的制备方法与流程
本发明属于高分子材料技术领域,尤其涉及一种光修复性偶氮苯聚合物的制备方法。
背景技术:
聚合物及其复合材料在长期的使用过程中会遭到化学侵蚀,机械性擦伤或碰撞以及热分解作用等环境的影响导致产生微裂缝,导致材料最终破坏,使得其寿命减少。这也是该材料在使用中性能劣化的直接原因。传统的修复方法如焊接、黏接、以及缝补仅仅是在宏观层面上将聚合物材料重新结合或增强破坏的部分。将自修复功能引入聚合物材料,使得材料内部产生损伤后能够自主修复,有助于得到使用寿命更长、性能更可靠和更经济的结构材料。如在微电子聚合物器件和胶粘剂使用中,由于热和力学疲劳产生的微裂缝导致的性能丧失是长期存在的问题,将自修复功能引入这些材料,可以大大提高微电子产品的可靠性和使用寿命。自修复材料的研究有助于得到具有仿生效果的材料,在生物医疗领域有应用前景,可以得到更耐久的人体关节。自修复材料的研究有助于开发特殊用途的材料,如在一定条件下可以恢复界面性能、导电和导热等性能的材料,对于实现界面性能或者传导性能的恢复,微米和亚微米级缝隙的自修复的研究更令人关注。这在航空航天等领域显得尤为重要。
几十年来自修复材料一直处于多样化的发展,按照是否需要外界因素来促进修复过程的进行可以划分为自动修复体系和非自动修复体系。自动修复体系以微胶囊体系为典型代表,主要是指在材料内部加入了一定量的单体(修复剂)和引发剂(催化剂),两者被隔开分散在材料内部,一旦材料出现裂痕,微胶囊也随之破裂,单体和引发剂在裂痕内混合并迅速发生聚合反应,从而在材料断裂所产生的裂痕处形成新的聚合物,完成对材料的修复。这种修复方法简单、修复效率高,但是其存在的最大缺陷是只能实现一次修复,并且grubb催化剂价格昂贵同时存在失活的可能性。随后white等人又开发了具有三维网络结构的微脉管自修复体系,这一体系可以实现多次循环修复,但其制备过程比较繁琐且价格昂贵。二烯类化合物主要利用聚合物材料内部本身具有的可逆化学反应来实现材料的自修复,并且可以在材料的同一位置进行多次循环修复,但其修复过程必须有外界条件的刺激才能进行,如光、热以及化学作用等。根据可逆化学键的种类又可分为两大类:(1)可逆非共价键:氢键作用、π-π堆积作用金属-配体的配位作用;(2)可逆共价键:diels-alder(da)反应加成物。diels-alder是典型的可逆反应。diels-alder反应条件温和、产率高、不需要催化剂、副反应少、具有可逆特性,可以用来制备修复聚合物材料。
偶氮苯化合物是指芳香环通过氮氮双键连接而成的化合物。在紫外光或热的作用下,偶氮苯发生可逆的顺反异构。在紫外光的照射下,偶氮苯的碳距离尺寸从反式的
通过掺杂或化学键将偶氮苯基团引入聚合物中,形成偶氮苯聚合物。相比于有机小分子,偶氮苯聚合物在力学性能、加工性能、热稳定性、成膜性等方面具有显著优点。偶氮苯聚合物具有光致顺反异构化性能、优异的力学性能和加工性能,在分子开关、液晶材料、光驱动等领域的应用潜力很大。
技术实现要素:
虽然偶氮苯聚合物具有光异构化作用,光作用下聚合物链具有一定迁移,但不足以其破损的表面具有修复功能。而蒽基团是三苯相连形成超共轭体系,因此,有很明显紫外吸收峰、强的荧光发射以及高的光反应活性。因此,本发明将蒽结构引入到偶氮苯聚合物侧链中,制备含蒽偶氮苯聚合物,结合蒽基团在光(365nm和254nm)激发条件下发生可逆diels-alder(da)加成反应,偶氮苯在紫外光和可见光激发下发生可逆的顺反异构行为,应用两种光响应结构的协同,制备出一种新型的光修复性偶氮苯聚合物。
首先,通过多步反应方法合成含蒽和偶氮苯基团单体,4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)。然后,再将mma-azo-an作为单体,偶氮二异丁腈(aibn)或过氧化二苯甲酰(bpo)作为引发剂,进行常规自由基聚合制备出聚4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(pmma-azo-an)。
蒽基团最主要的光化学反应是在长波长紫外光(365nm)激发条件下的[4+4]光二聚反应,是一种紫外光diels-alder(da)加成反应,所得到的二聚体可以在短波长(254nm)的条件下还原生成蒽基团。通过这样的光二聚反应用来在聚合物光交联与解交联基团,可以用于修复材料中光修复基团。
蒽的可逆反应反应式如下:
本发明采用如下技术方案为:
一种光修复性偶氮苯聚合物(pmma-azo-an)的结构通式如下:
式中:n取整数,分子量为3000~100000,pdi=1.3~2.5。
所述的pmma-azo-an是通过4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)常规自由基聚合得到,偶氮二异丁腈(aibn)或过氧化二苯甲酰(bpo)作为引发剂,溶剂可以为四氢呋喃(thf)、n,n-二甲基甲酰胺、丙酮、苯甲醚、甲苯等良溶剂,聚合温度为50~100℃,aibn或bpo用量为单体摩尔数的1%,反应结束后纯化获得pmma-azo-an。
所述的4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an),其结构通式如下:
所述的mma-azo-an的制备是通过对硝基苯酚的醚化、硝基还原、重氮偶合、酯化、取代、再酯化等多步反应获得的,其反应方程为:
mma-azo-an的制备方法为:首先制备4-羟基己氧基偶氮苯酚;然后,制备2-溴乙酸蒽-10-基甲基酯;最后将4-羟基己氧基偶氮苯酚与2-溴乙酸蒽-10-基甲基酯取代制得4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯、再经酯化反应制得含蒽和偶氮苯基团单体,4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)。
所述的4-羟基己氧基偶氮苯酚的制备方法为:
(1)将6-氯己醇与对硝基苯酚,在无机碱存在的条件下,以ki作为催化剂,反应制得中间体对硝基苯氧基己醇,其中,6-氯己醇与对硝基苯酚的摩尔比为0.5-2:1,所述无机碱为k2co3或na2co3,其用量为对硝基苯酚摩尔量的1.24倍;ki用量为对硝基苯酚质量的0.08%,反应条件为:以dmf作为溶剂,120℃回流反应6h后停止,乙醇重结晶制得中间体;
(2)对硝基苯氧基己醇在还原剂fe/nh4cl存在的条件下还原制得中间体对氨基苯氧基己醇;其中,还原剂fe的用量为对硝基苯氧基己醇摩尔量的1-10倍,fe和nh4cl的摩尔比为1:1~3;
(3)在氧化剂亚硝酸钠作用下,对氨基苯氧基己醇进行重氮化,与苯酚偶合反应制备出中间体4-羟基己氧基偶氮苯酚;其中,对氨基苯氧基己醇与苯酚的摩尔比为1:1~2,所述亚硝酸钠的用量为苯酚摩尔量的1~2倍,0~5℃进行重氮偶合反应,乙醇重结晶,干燥得到中间体;
所述的2-溴乙酸蒽-10-基甲基酯的制备方法为:蒽甲醇与溴乙酰溴在缚酸剂存在的条件下,0~5℃下酯化制得中间体2-溴乙酸蒽-10-基甲基酯;其中,蒽甲醇与溴乙酰溴的摩尔比为1:1~1.5,三乙胺作为缚酸剂,其用量是溴乙酰溴摩尔量的1~1.5倍。
所述的4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的制备方法为:2-溴乙酸蒽-10-基甲基酯与4-羟基己氧基偶氮苯酚取代反应制得中间体4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯;其中,2-溴乙酸蒽-10-基甲基酯与4-羟基己氧基偶氮苯酚摩尔比为1~1.5:1,反应条件为60℃回流反应5h,乙醇重结晶制得。
所述的4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)的制备方法为:将4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯与甲基丙烯酰氯低温下酯化制得;其中,4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯与甲基丙烯酰氯的摩尔比为1:1~1.5,酯化反应条件为0~5℃条件下,通过恒压滴液漏斗将甲基丙烯酰氯缓慢滴入反应液中,5h后乙醇重结晶制得。
有益效果:
本发明制备出了含蒽偶氮苯高分子材料,蒽结构在光辐照下具有优异光交联和解交联特点,偶氮苯在光辐照下具有优异的光异构下特性,伴随着聚合链迁移性,通过光辐射,聚合物膜发生偶氮苯的异构化和蒽环的加成的协同效应,对破碎的聚合物表面具有光修复性能。
附图说明
图1、经过提纯的对硝基苯氧基己醇的核磁氢谱图(cdcl3为溶剂);
图2、经过提纯的对氨基苯氧基己醇的核磁氢谱图(氘代dmso为溶剂);
图3、经过提纯的4-羟基己氧基偶氮苯酚的核磁氢谱图(氘代dmso为溶剂);
图4、经过提纯的2-溴乙酸蒽-10-基甲基酯的核磁氢谱图(cdcl3为溶剂);
图5、经过提纯的4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的核磁氢谱图(cdcl3为溶剂);
图6、经过提纯的4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)的高效液相色谱图;
图7、经过提纯的4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)的核磁氢谱图(cdcl3为溶剂);
图8、聚合物pmma-azo-an(mn=3.18×104g/mol,pdi=1.86)的核磁氢谱图。
图9、聚合物pmma-azo-an(mn=3.18×104g/mol,pdi=1.86)的分子量微分分布图。
具体实施方式
下面结合实施例对本发明做进一步说明:
实施例1
对硝基苯氧基己醇的合成,由6-氯己醇与对硝基苯酚反应制得。
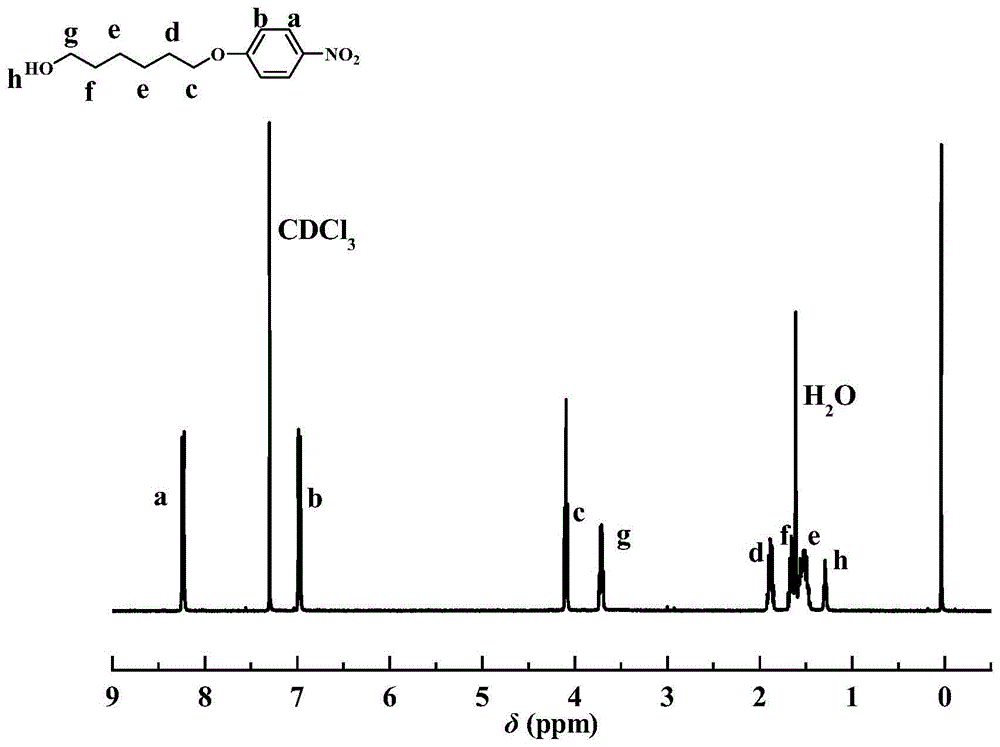
将6-氯己醇(37.0mmol,5.05g)、4-硝基酚(44.4mmol,6.18g)、k2co3(55.4mmol,7.65g)、ki(5mg)、dmf(50ml)加入三口烧瓶中,120℃回流,反应6h后停止反应,将混合溶液倒入大量水中,得到米白色沉淀,过滤乙醇重结晶,干燥后,得到对硝基苯氧基己醇,产率约87%,高效液相色谱分析纯度为95%。
图1为经过提纯的对硝基苯氧基己醇的核磁氢谱图(cdcl3为溶剂)。化学位移8.08ppm和6.99ppm的峰对应于苯环碳上的质子峰(a,b),化学位移4.08ppm的峰对应于与氧相连的亚甲基上的质子峰(c),化学位移3.72ppm的峰对应于与羟基相连的亚甲基的质子峰(g),化学位移1.89ppm、1.66ppm和1.51ppm的峰为亚甲基的质子峰(d,f,e),化学位移1.29ppm的峰为羟基的质子峰(h)。对a、b、c、d、e、f、g、h处的峰面积进行积分,面积比为2.00:2.06:2.12:2.18:4.24:2.08:2.08:1.02,这与理论值吻合,证明成功制备出对硝基苯氧基己醇。
实施例2
对硝基苯氧基己醇的合成
将6-氯己醇(22.2mmol,3.03g)、4-硝基酚(44.4mmol,6.18g)、k2co3(55.4mmol,7.65g)、ki(5mg)、dmf(50ml)加入三口烧瓶中,120℃回流,反应6h后停止反应,将混合溶液倒入大量水中,得到米白色沉淀,过滤乙醇重结晶,干燥后,得到对硝基苯氧基己醇,产率约89%,高效液相色谱分析纯度为95%。
实施例3
对硝基苯氧基己醇的合成
将6-氯己醇(88.8mmol,12.12g)、4-硝基酚(44.4mmol,6.18g)、k2co3(55.4mmol,7.65g)、ki(5mg)、dmf(50ml)加入三口烧瓶中,120℃回流,反应6h后停止反应,将混合溶液倒入大量水中,得到米白色沉淀,过滤乙醇重结晶,干燥后,得到对硝基苯氧基己醇,产率约85%,高效液相色谱分析纯度为96%。
实施例4
对氨基苯氧基己醇的合成,对硝基苯氧基己醇还原反应制得。
将氯化铵(83.7mmol,4.48g)溶于去离子水(40ml)中,在三口烧瓶中加入铁粉(50.5mmol,2.80g)。将对硝基苯氧基己醇(16.7mmol,4.0g),溶于140ml甲醇中,然后加入到快速搅拌的三口烧瓶中,进行硝基还原反应。室温反应3h后于50℃反应,跟踪反应进程。反应完毕,过滤除去铁粉,旋蒸除去甲醇,抽滤掉去离子水,乙醇重结晶,干燥,得到对氨基苯氧基己醇,高效液相色谱分析纯度为95%,产率约为65%。
图2为经过提纯的对氨基苯氧基己醇的核磁氢谱图(氘代dmso为溶剂)。化学位移6.52ppm和6.64ppm的峰对应于苯环碳上的质子峰(a,b),化学位移3.79ppm的峰对应于与氧相连的亚甲基的质子峰(c),化学位移3.39ppm的峰对应于与羟基相连的亚甲基的质子峰(d),化学位移1.65ppm、1.44ppm和1.36ppm的峰为亚甲基的质子峰(e,f,g),化学位移4.68ppm的峰为氨基的质子峰(h),化学位移4.37ppm的峰为羟基上氢的特征吸收峰(i)。对a、b、c、d、e、f、g、h、i处的峰面积积分,面积比为1.92:1.91:1.94:1.83:1.97:2.05:4.06:1.83:1.00,这与理论值吻合,证明成功制备出对氨基苯氧基己醇。
实施例5
对氨基苯氧基己醇的合成,对硝基苯氧基己醇还原反应制得。
将氯化铵(16.7mmol,0.90g)溶于去离子水(40ml)中,在三口烧瓶中加入铁粉(16.7mmol,0.56g)。将对硝基苯氧基己醇(16.7mmol,4.0g),溶于140ml甲醇中,然后加入到快速搅拌的三口烧瓶中,进行硝基还原反应。室温反应3h后于50℃反应,跟踪反应进程。反应完毕,过滤除去铁粉,旋蒸除去甲醇,抽滤掉去离子水,乙醇重结晶,干燥,得到对氨基苯氧基己醇,高效液相色谱分析纯度为95%,产率约为45%。
实施例6
对氨基苯氧基己醇的合成,对硝基苯氧基己醇还原反应制得的。
将氯化铵(501.0mmol,27.0g)溶于去离子水(80ml)中,在三口烧瓶中加入铁粉(167.0mmol,5.6g)。将对硝基苯氧基己醇(16.7mmol,4.0g),溶于240ml甲醇中,然后加入到快速搅拌的三口烧瓶中,进行硝基还原反应。室温反应3h后于50℃反应,跟踪反应进程。反应完毕,过滤除去铁粉,旋蒸除去甲醇,抽滤掉去离子水,乙醇重结晶,干燥,得到对氨基苯氧基己醇,高效液相色谱分析纯度为95%,产率约为75%。
实施例7
4-羟基己氧基偶氮苯酚的合成,对氨基苯氧基己醇重氮偶合反应制得。
三口烧瓶中加入对氨基苯氧基己醇(9.0mmol,2.6g)。将盐酸(2.7ml)加入水(8.0ml)中稀释,然后倒入三口烧瓶中。将nano2(9.0mmol,0.62g)的水溶液通过恒压滴液漏斗缓慢滴入三口烧瓶中,冰浴(0~5℃)条件下,进行重氮化反应。反应完后,过滤滴入含苯酚(9.0mmol,0.84g)、naoh(10.8mmol,0.43g)、na2co3(17.8mmol,1.89g)、nahco3(17.8mmol,1.50g)的水溶液中,进行偶合反应,反应过程中调节ph=8-9。反应结束后,抽滤,乙醇重结晶,干燥得到4-羟基己氧基偶氮苯酚。高效液相色谱分析纯度为95%,产率约为70%。
图3是经过提纯的4-羟基己氧基偶氮苯酚的核磁氢谱图(氘代dmso为溶剂),化学位移7.80ppm、7.75ppm、7.11ppm、6.93ppm处的峰为苯环上氢的特征吸收峰(g,h,f,i),化学位移4.07ppm的峰为与氧相连的亚甲基的质子峰(e),化学位移3.41ppm的峰为与羟基相连的亚甲基的质子峰(a),化学位移1.70ppm、1.45ppm、1.40ppm处的峰是亚甲基的质子峰(d,b,c),化学位移4.37ppm、10.20ppm处的峰是羟基的质子峰(k,j)。对a、b、c、d、e、f、g、h、i、j、k处的峰面积积分,面积比为1.96:1.96:3.62:2.05:1.91:1.83:1.74:1.76:1.79:0.95:1.00,与理论值吻合,证明成功制备出4-羟基己氧基偶氮苯酚。
实施例8
4-羟基己氧基偶氮苯酚的合成,对氨基苯氧基己醇重氮偶合反应制得。
三口烧瓶中加入对氨基苯氧基己醇(9.0mmol,2.6g)。将盐酸(2.7ml)加入水(8.0ml)中稀释,然后倒入三口烧瓶中。将nano2(13.5mmol,0.93g)的水溶液通过恒压滴液漏斗缓慢滴入三口烧瓶中,冰浴(0~5℃)条件下,进行重氮化反应。反应完后,过滤滴入含苯酚(13.5mmol,1.26g)、naoh(16.2mmol,0.64g)、na2co3(17.8mmol,1.89g)、nahco3(17.8mmol,1.50g)的水溶液中,进行偶合反应,反应过程中调节ph=8-9。反应结束后,抽滤,乙醇重结晶,干燥得到4-羟基己氧基偶氮苯酚。高效液相色谱分析纯度为96%,产率约为72%。
实施例9
4-羟基己氧基偶氮苯酚的合成,对氨基苯氧基己醇重氮偶合反应制得的。
三口烧瓶中加入对氨基苯氧基己醇(9.0mmol,2.6g)。将盐酸(2.7ml)加入水(8.0ml)中稀释,然后倒入三口烧瓶中。将nano2(18.0mmol,12.4g)的水溶液通过恒压滴液漏斗缓慢滴入三口烧瓶中,冰浴(0~5℃)条件下,进行重氮化反应。反应完后,过滤滴入含苯酚(18.0mmol,1.68g)、naoh(21.6mmol,0.86g)、na2co3(17.8mmol,1.89g)、nahco3(17.8mmol,1.50g)的水溶液中,进行偶合反应,反应过程中调节ph=8-9。反应结束后,抽滤,乙醇重结晶,干燥得到4-羟基己氧基偶氮苯酚。高效液相色谱分析纯度为95%,产率约为75%。
实施例10
2-溴乙酸蒽-10-基甲基酯的合成,蒽甲醇与溴乙酰溴低温下酯化制得。
将溴乙酰溴(11.5mmol,1.0ml)溶于10ml二氯甲烷,在冰浴(0~5℃)条件下,通过恒压滴液漏斗缓慢滴入盛有蒽甲醇(9.7mmol,2.01g)和三乙胺(12.5mmol,1.26g)的四氢呋喃溶液(10ml)的圆底烧瓶中,反应结束后洗涤,干燥,乙醇重结晶,得到2-溴乙酸蒽-10-基甲基酯。高效液相色谱分析纯度为95%,产率约为60%。
如图4是经过提纯的2-溴乙酸蒽-10-基甲基酯的核磁氢谱图,对于此谱图,化学位移δ8.54ppm、8.31ppm、8.06ppm、7.58ppm和7.51ppm处的峰对应于蒽环上的碳的质子峰(a~e),化学位移δ6.27ppm处对应于与蒽环连接的碳的质子峰,化学位移δ4.70ppm对应于与溴原子相连的碳的质子峰。通过a、b、c、d、e、f、g处的面积比为0.5:0.99:1.01:1.00:1.00:1.04:1.00,这与理论值相符合,成功制备出2-溴乙酸蒽-10-基甲基酯。
实施例11
2-溴乙酸蒽-10-基甲基酯的合成,蒽甲醇与溴乙酰溴低温下酯化制得。
将溴乙酰溴(9.7mmol,0.8ml)溶于10ml二氯甲烷,在冰浴(0~5℃)条件下,通过恒压滴液漏斗缓慢滴入盛有蒽甲醇(9.7mmol,2.01g)和三乙胺(9.7mmol,0.98g)的四氢呋喃溶液(10ml)的圆底烧瓶中,反应结束后洗涤,干燥,乙醇重结晶,得到2-溴乙酸蒽-10-基甲基酯。高效液相色谱分析纯度为96%,产率约为56%。
实施例12
2-溴乙酸蒽-10-基甲基酯的合成,蒽甲醇与溴乙酰溴低温下酯化制得。
将溴乙酰溴(14.5mmol,1.3ml)溶于10ml二氯甲烷,在冰浴(0~5℃)条件下,通过恒压滴液漏斗缓慢滴入盛有蒽甲醇(9.7mmol,2.01g)和三乙胺(14.5mmol,1.46g)的四氢呋喃溶液(10ml)的圆底烧瓶中,反应结束后洗涤,干燥,乙醇重结晶,得到2-溴乙酸蒽-10-基甲基酯。高效液相色谱分析纯度为95%,产率约为63%。
实施例13
4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的合成,由4-羟基己氧基偶氮苯酚与2-溴乙酸蒽-10-基甲基酯取代反应制得。
将4-羟基己氧基偶氮苯酚(3.8mmol,1.19g)、2-溴乙酸蒽-10-基甲基酯(3.9mmol,1.29g)、k2co3(4.5mmol,0.62g)、丙酮(30ml)依次加入圆底烧瓶中,于60℃回流反应5h。反应结束后过滤除去无机盐,旋蒸,乙醇重结晶,得到产物4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的偶氮苯。高效液相色谱分析纯度为95%,产率约为70%。
如图5是4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的核磁氢谱图,对于此谱图,化学位移δ8.53ppm、8.32ppm、8.05ppm、7.57ppm和7.50ppm处的峰对应于蒽环上的碳的质子峰(a~e),化学位移δ6.32ppm处对应于与蒽环连接的碳的质子峰(f),化学位移δ4.70ppm对应于与酯基相连的碳的质子峰(g),化学位移δ7.86ppm和6.98ppm对应于偶氮苯苯环上面的碳的质子峰(h,i),化学位移δ4.04ppm、3.67ppm、1.63ppm、1.50ppm和1.84ppm(j~o)对应于偶氮苯与甲基丙烯酸酯中间6个碳的质子峰。最后化学位移δ1.25ppm处对应于端羟基的质子吸收峰。通过a、b、c、d、e、f、g、h、i、j、k、l、m、n、o处面积比为1.00:0.92:0.95:0.98:0.50:1.00:1.09:1.95:1.95:1.05:1.02:2.16:0.97:1.09:0.55这与理论值相符合,成功制备出4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯。
实施例14
4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的合成,由4-羟基己氧基偶氮苯酚与2-溴乙酸蒽-10-基甲基酯取代反应制得。
将4-羟基己氧基偶氮苯酚(3.8mmol,1.19g)、2-溴乙酸蒽-10-基甲基酯(4.6mmol,1.75g)、k2co3(5.0mmol,0.69g)、丙酮(30ml)依次加入圆底烧瓶中,于60℃回流反应5h。反应结束后过滤除去无机盐,旋蒸,乙醇重结晶,得到产物4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的偶氮苯。高效液相色谱分析纯度为95%,产率约为75%。
实施例15
4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的合成,由4-羟基己氧基偶氮苯酚与2-溴乙酸蒽-10-基甲基酯取代反应制得。
将4-羟基己氧基偶氮苯酚(3.8mmol,1.19g)、2-溴乙酸蒽-10-基甲基酯(5.7mmol,1.88g)、k2co3(6.0mmol,0.83g)、丙酮(30ml)依次加入圆底烧瓶中,于60℃回流反应5h。反应结束后过滤除去无机盐,旋蒸,乙醇重结晶,得到产物4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯的偶氮苯。高效液相色谱分析纯度为95%,产率约为80%。
实施例16
单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)的合成,由4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯与甲基丙烯酰氯低温下酯化制得。
将4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯(0.74mmol,0.42g)、三乙胺(1.2mmol,0.12g)溶于10ml四氢呋喃中配成反应液于圆底烧瓶中。将甲基丙烯酰氯(1.0mmol,0.1ml)溶于10ml四氢呋喃中,在冰浴(0~5℃)条件下,通过恒压滴液漏斗缓慢滴入反应液中。5h反应结束后,洗涤,干燥,旋蒸,乙醇重结晶,得到单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)。图6所示,高效液相色谱得到纯度为96.3%,产率约为60%。
图7是单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)的核磁氢谱图,对于此谱图,化学位移δ8.52ppm、8.29ppm、8.02ppm、7.56ppm和7.49ppm处的峰对应于蒽环上的碳的质子峰(a~e),化学位移δ6.31ppm处对应于与蒽环连接的碳的质子峰(f),化学位移δ4.70ppm对应于与酯基相连的碳的质子峰(g),化学位移δ7.81ppm和6.96ppm对应于偶氮苯苯环上面的碳的质子峰(h,i),化学位移δ4.14ppm、4.02ppm、1.49ppm、1.72ppm和1.80ppm对应于偶氮苯与甲基丙烯酸酯中间6个碳的质子峰(j~n)。化学位移δ1.94ppm处对应于连在碳碳双键上甲基的质子峰(q),最后化学位移δ5.54ppm和6.09ppm处对应于碳碳双键上两个不同位的质子峰(o,p)。通过a、b、c、d、e、f、g、h、i、j、k、l、m、n、o、p、q处面积比为:2.05:2.08:2.06:2.10:1.03:2.09:1.89:3.79:4.13:1.82:1.83:3.90:2.39:2.09:2.94:1:00:1,这与理论值相符合,成功制备出4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯。
实施例17
单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)的合成,由4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯与甲基丙烯酰氯低温下酯化制得。
将4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯(0.74mmol,0.42g)、三乙胺(1.0mmol,0.1g)溶于10ml四氢呋喃中配成反应液于圆底烧瓶中。将甲基丙烯酰氯(0.74mmol,0.07ml)溶于10ml四氢呋喃中,在冰浴(0~5℃)条件下,通过恒压滴液漏斗缓慢滴入反应液中。5h反应结束后,洗涤,干燥,旋蒸,乙醇重结晶,得到单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)。图6所示,高效液相色谱得到纯度为96%,产率约为50%。
实施例18
单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)的合成,由4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯与甲基丙烯酰氯低温下酯化制得。
将4’-(羟己氧基)-4-(氧乙酸蒽甲酯基)偶氮苯(0.74mmol,0.42g)、三乙胺(1.2mmol,0.12g)溶于10ml四氢呋喃中配成反应液于圆底烧瓶中。将甲基丙烯酰氯(1.11mmol,0.105ml)溶于10ml四氢呋喃中,在冰浴(0~5℃)条件下,通过恒压滴液漏斗缓慢滴入反应液中。5h反应结束后,洗涤,干燥,旋蒸,乙醇重结晶,得到单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an)。图6所示,高效液相色谱得到纯度为96%,产率约为62%。
实施例19
聚合物聚4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(pmma-azo-an)的合成。
以偶氮二异丁腈(aibn)为引发剂,n,n-二甲基甲酰胺(dmf)为溶剂,将单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an,0.63g,1.0mmol),引发剂aibn为单体摩尔数的1%,dmf(5ml),加入聚合瓶中,液氮冷冻抽真空,最后充入氩气,于50℃进行均聚。10h反应结束后,用四氢呋喃溶解,在乙醇中沉淀3次,用离心管离心后得到均聚物pmma-azo-an。
图8是为pmma-azo-an的核磁氢谱图。化学位移δ8.32ppm、8.17ppm、7.62ppm、7.43ppm、7.36ppm处对应于蒽环上的质子峰(c~g),化学位移δ6.13ppm处对应于与蒽环连接的碳的质子峰(h),化学位移δ4.44ppm对应于与酯基相连的碳的质子峰(i),化学位移δ7.84ppm、7.73ppm、6.83ppm和6.72ppm处对应于偶氮苯苯环上面的碳的质子峰(k,j),化学位移3.83ppm对应于偶氮苯上的氧元素相连的碳的质子峰(l),化学位移δ3.92ppm对应于与酯基上的氧元素相连的碳的质子峰(m)。积分所得各峰面积比值也与理论值基本相吻合,证明成功合成了均聚物pmma-azo-an。
图9是聚合物mma-azo-an的分子量微分分布图,图中峰型为单峰型,是一种单一组成聚合物,积分得到聚合物数均分子量为31800g/mol,pdi为1.86。
实施例20
聚合物聚4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(pmma-azo-an)的合成。
以偶氮二异丁腈(aibn)为引发剂,苯甲醚为溶剂,将单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an,0.63g,1.0mmol)为,引发剂aibn为单体摩尔数的1%,苯甲醚(5ml),加入聚合瓶中,液氮冷冻抽真空,最后充入氩气,于100℃进行均聚。10h反应结束后,用四氢呋喃溶解,在乙醇中沉淀3次,用离心管离心后得到均聚物pmma-azo-an。
实施例21
聚合物聚4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(pmma-azo-an)的合成。
以偶氮二异丁腈(aibn)为引发剂,四氢呋喃为溶剂,将单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an,0.63g,1.0mmol)为,引发剂aibn为单体摩尔数的1%,四氢呋喃(5ml),加入聚合瓶中,液氮冷冻抽真空,最后充入氩气,于70℃进行均聚。10h反应结束后,用四氢呋喃溶解,在乙醇中沉淀3次,用离心管离心后得到均聚物pmma-azo-an。
实施例22
聚合物聚4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(pmmaazo-an)的合成。
以偶氮二异丁腈(aibn)为引发剂,丙酮为溶剂,将单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an,0.63g,1.0mmol)为,引发剂aibn为单体摩尔数的1%,丙酮(5ml),加入聚合瓶中,液氮冷冻抽真空,最后充入氩气,于60℃进行均聚。10h反应结束后,用四氢呋喃溶解,在乙醇中沉淀3次,用离心管离心后得到均聚物pmma-azo-an。
实施例23
聚合物聚4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(pmmaazo-an)的合成。
以偶氮二异丁腈(aibn)为引发剂,甲苯为溶剂,将单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an,0.63g,1.0mmol)为,引发剂aibn为单体摩尔数的1%,甲苯(5ml),加入聚合瓶中,液氮冷冻抽真空,最后充入氩气,于90℃进行均聚。10h反应结束后,用四氢呋喃溶解,在乙醇中沉淀3次,用离心管离心后得到均聚物pmma-azo-an。
实施例24
聚合物聚4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(pmma-azo-an)的合成。
以过氧化二苯甲酰(bpo)为引发剂,苯甲醚为溶剂,将单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an,0.63g,1.0mmol)为,引发剂bpo为单体摩尔数的1%,苯甲醚(5ml),加入聚合瓶中,液氮冷冻抽真空,最后充入氩气,于100℃进行均聚。10h反应结束后,用四氢呋喃溶解,在乙醇中沉淀3次,用离心管离心后得到均聚物pmma-azo-an。
实施例25
聚合物聚4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(pmma-azo-an)的合成。
以过氧化二苯甲酰(bpo)为引发剂,甲苯为溶剂,将单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo-an,0.63g,1.0mmol)为,引发剂bpo为单体摩尔数的1%,甲苯(5ml),加入聚合瓶中,液氮冷冻抽真空,最后充入氩气,于90℃进行均聚。10h反应结束后,用四氢呋喃溶解,在乙醇中沉淀3次,用离心管离心后得到均聚物pmma-azo-an。
实施例25
聚合物pmma-azo-an(0.1g)溶解于三氯甲烷(5ml)中,基材为pet膜,旋涂在pet上成膜,置于80℃鼓风烘箱5h,用刀片在聚合物薄膜上划痕,紫外光辐射于薄膜10min进行光修复,划痕消失显示优异的光修复性。
对照例1
聚合物聚4-(甲基丙烯酸己酯氧基)偶氮苯(pmma-azo)的合成。
以过氧化二苯甲酰(bpo)为引发剂,甲苯为溶剂,将单体4’-(甲基丙烯酸己酯氧基)-4-(氧乙酸蒽-2-甲酯基)偶氮苯(mma-azo,0.36g,1.0mmol)为,引发剂bpo为单体摩尔数的1%,甲苯(5ml),加入聚合瓶中,液氮冷冻抽真空,最后充入氩气,于90℃进行均聚。10h反应结束后,用四氢呋喃溶解,在乙醇中沉淀3次,用离心管离心后得到均聚物pmma-azo。
聚合物pmma-azo(0.1g)溶解于三氯甲烷(5ml)中,基材为pet膜,旋涂在pet上成膜,置于80℃鼓风烘箱5h,用刀片在聚合物薄膜上划痕,紫外光辐射于薄膜10min进行光辐射,划痕无明显变化,通过对照例,表明偶氮苯和蒽结构在光辐射下具有协同效应,才能通过光修复表面划痕。
以上所述仅为本发明优选的实施例而已,并不用于限制本发明,对本领域的技术人员来说,其依然可以对前述各实施例中所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!