氮杂环羧酸中间体及氮杂环丁烷-3-羧酸的制备方法与流程
本发明涉及医药化工技术领域,且特别涉及氮杂环羧酸中间体及氮杂环丁烷-3-羧酸的制备方法。
背景技术:
多发性硬化症是最常见的一种中枢神经脱髓鞘疾病,由于炎症和组织损失原因,大脑、视神经和脊髓的正常运作遭到免疫系统的破坏。发病时,病人的免疫系统会异常攻击大脑、脊髓和视神经等神经细胞的髓鞘——它们包裹神经细胞起绝缘和支撑作用。这种神经组织病变引起的炎症和随后的组织损害可能会导致一系列的症状,包括肌肉无力、疲劳、视觉问题,并最终可能导致残疾。目前,全世界范围约有250万患者,大多数多发性硬化症患者经历首次症状约在20和40岁之间,使得该病成为青壮年人群中因非外伤致残的主要原因之一。
辛波莫德是novartis开发用于持续延缓继发性多发性硬化症(spms)患者的药物。siponimod于2019年3月26日被美国fda批准上市,而氮杂环丁烷-3-羧酸是辛波莫德(siponimod)的重要中间体。
目前,文献公开的氮杂环丁烷-3-羧酸的制备方法最早来自专利us20100249399中的制备方法,其合成路线为:
该合成路线中由1-二苯甲基-3-羟基氮杂环丁烷为起始原料通过4步反应得到氮杂环丁烷-3-羧酸,合成过程中采用了剧毒试剂氰化钠,大大增加了生产过程中的危险性,导致操作困难,不利于常规生产。
与us20100249399相似,现有技术中用于合成氮杂环丁烷-3-羧酸的工艺均存在合成路线长、合成成本高的问题,部分方法还存在危险系数大的问题。鉴于此,特提出本发明。
技术实现要素:
本发明的目的在于提供一种用于制备氮杂环丁烷-3-羧酸的氮杂环羧酸中间体的制备方法,旨在缩短氮杂环丁烷-3-羧酸制备的反应路线,降低反应的危险性,适于工业化生产。
本发明的另一目的在于提供一种氮杂环丁烷-3-羧酸的制备方法,其工艺路线短、制备成本低、操作安全。
本发明解决其技术问题是采用以下技术方案来实现的。
本发明提出了一种用于制备氮杂环丁烷-3-羧酸的氮杂环羧酸中间体的制备方法,包括如下步骤:
将氮杂环化合物和wittig反应试剂混合在0-10℃的条件下进行wittig反应并分离得到氮杂环甲醛中间体;
将氮杂环甲醛中间体进行氧化反应并分离得到氮杂环羧酸中间体;
其中,氮杂环化合物选自1-二苯甲基-3-氮杂环丁酮、1-苄基氮杂环丁烷-3-酮、1-boc氮杂环丁烷-3-酮和1-乙酰基氮杂啶-3-酮中的至少一种;优选为1-二苯甲基-3-氮杂环丁酮;
优选地,wittig反应试剂包括甲氧基甲基三苯基氯化膦和甲基三苯基溴化膦中的至少一种。
本发明还提出一种氮杂环丁烷-3-羧酸的制备方法,包括如下步骤:
采用上述制备方法制备氮杂环羧酸中间体;
将氮杂环羧酸中间体进行氢解。
本发明实施例提供一种用于制备氮杂环丁烷-3-羧酸的氮杂环羧酸中间体的制备方法的有益效果是:其以1-二苯甲基-3-氮杂环丁酮、1-苄基氮杂环丁烷-3-酮、1-boc氮杂环丁烷-3-酮、1-乙酰基氮杂啶-3-酮等氮杂环化合物为原料,通过与甲氧基甲基三苯基氯化膦在0-10℃的条件下进行wittig反应制备得到氮杂环甲醛中间体,再将氮杂环甲醛中间体进行氧化后得到氮杂环羧酸中间体。通过两步反应制备得到氮杂环羧酸中间体,缩短了反应路线,具有原料易得、操作简单安全、方法可靠、成本低、适于工业化生产等优点。
本发明还提供了一种氮杂环丁烷-3-羧酸的制备方法,应用上述制备方法制备氮杂环羧酸中间体,然后经过氢解步骤制备得到氮杂环丁烷-3-羧酸,工艺路线短、原料易得,且操作的安全性高。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
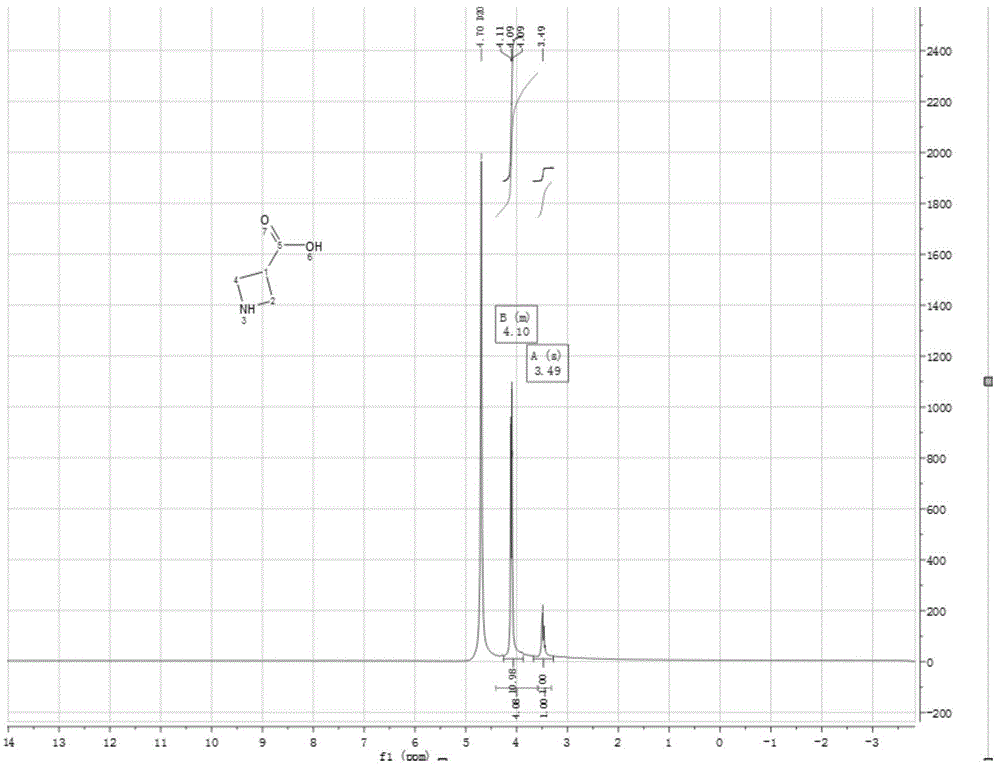
图1为本发明实施例制备得到产品的核磁共振氢谱图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
下面对本发明实施例提供的氮杂环羧酸中间体及氮杂环丁烷-3-羧酸的制备方法进行具体说明。
本发明实施例提供的一种氮杂环丁烷-3-羧酸的制备方法,其包括:
需要说明的是,以1-二苯甲基-3-氮杂环丁酮、1-苄基氮杂环丁烷-3-酮、1-boc氮杂环丁烷-3-酮、1-乙酰基氮杂啶-3-酮等氮杂环化合物为原料(上述化学式仅表示了1-二苯甲基-3-氮杂环丁酮为原料的反应原理),通过与甲氧基甲基三苯基氯化膦进行wittig反应制备得到氮杂环甲醛中间体,再将氮杂环甲醛中间体进行氧化后得到氮杂环羧酸中间体,再经过氢解制备得到氮杂环丁烷-3-羧酸。
整体步骤而言,氮杂环丁烷-3-羧酸的制备方法包括氮杂环羧酸中间体的制备和氢解过程,具体步骤如下:
s1、wittig反应
将氮杂环化合物和wittig反应试剂混合进行wittig反应并分离得到氮杂环甲醛中间体;其中,氮杂环化合物选自1-二苯甲基-3-氮杂环丁酮、1-苄基氮杂环丁烷-3-酮、1-boc氮杂环丁烷-3-酮和1-乙酰基氮杂啶-3-酮中的至少一种;优选为1-二苯甲基-3-氮杂环丁酮。wittig反应试剂包括甲氧基甲基三苯基氯化膦和甲基三苯基溴化膦中的至少一种。1-苄基氮杂环丁烷-3-酮、1-boc氮杂环丁烷-3-酮和1-乙酰基氮杂啶-3-酮中的化学式如下:
需要说明的是,发明人创造性地采用以上几种氮杂环化合物为原料进行wittig反应,可以经过三步反应制备得到氮杂环丁烷-3-羧酸,缩短合成路线和合成成本,而且原料易得,操作安全可靠,适合于工业化生产。本发明实施例中提及的四种氮杂环化合物均为市购原料,原料易得,且价格低廉。
优选地,wittig反应是在碱性条件下进行,优选地ph为9-10。发明人发现,wittig反应在碱性条件下能够保证氮杂环甲醛中间体的收率,若出现过酸或过碱的条件均会影响反应进行,若碱性较弱可能无法引发反应,若碱性条件过强可能也会导致氮杂环甲醛中间体收率低。
进一步地,wittig反应中所采用的碱性试剂为氢化钠、六甲基二硅基胺基锂和叔丁醇钾中的至少一种;优选为六甲基二硅基胺基锂。以上几种碱性试剂均适合应用于本发明实施例中的wittig反应体系,使反应控制在规定的ph范围内。甲氧基甲基三苯基氯化膦与碱性试剂的摩尔比为1:1.05-2.0;优选为1:1.2-1.6;更优选为1:1.5,通过调控碱性试剂的用量来精确控制反应的碱性条件,若碱性试剂的用量超出上述范围有可能导致反应无法进行或者中间体的收率较低。
进一步地,氮杂环化合物和甲氧基甲基三苯基氯化膦的反应温度为0-10℃,反应时间为3-12h;优选地,反应温度为0-5℃,反应时间为6-8h。wittig反应的反应温度和时间也是影响反应的重要因素,若反应温度过高或过低均有可能导致反应无法进行,反应温度控制在0-5℃为宜;反应时间主要是影响中间体的收率,若反应时间过短会导致最终产品的收率不理想。
优选地,在wittig反应中,氮杂环化合物和甲氧基甲基三苯基氯化膦的摩尔比为1:2.5-4.0;优选为1:3.0-3.2;更优选为1:3.0。在wittig反应中,氮杂环化合物和甲氧基甲基三苯基氯化膦控制在上述范围内为宜,以进一步提升中间体的收率,若甲氧基甲基三苯基氯化膦的用量过少会导致反应无法进行。
具体地,wittig反应的反应溶剂为n,n-二甲基甲酰胺和/或四氢呋喃;优选为四氢呋喃。在wittig反应中是将甲氧基甲基三苯基氯化膦溶解后先与碱性试剂混合搅拌1-3h,然后再滴加氮杂环化合物溶液进行反应;优选地,在wittig反应之后,进行萃取、浓缩。将甲氧基甲基三苯基氯化膦溶解后先与碱性试剂混合一段时间形成稳定的碱性环境,再缓慢滴加氮杂环化合物溶液进行反应,能够提升中间体的收率。
s2、氧化反应
将氮杂环甲醛中间体进行氧化反应并分离得到氮杂环羧酸中间体,通过将氮杂环甲醛上的醛基进行氧化得到羧基。
优选地,氧化反应中采用的氧化剂选自双氧水和/或亚氯酸钠;优选为亚氯酸钠。双氧水和亚氯酸钠均适合于本发明实施例中的反应体系,能够将醛基充分氧化。氮杂环甲醛中间体与氧化剂的摩尔比为1:1-1.5;优选为1:1.0-1.2;更优选为1:1.1。发明人通过进一步控制氮杂环甲醛中间体与氧化剂的用量,提升反应速率并使氮杂环甲醛中间体充分氧化,提高反应的收率。
优选地,氧化反应是将氮杂环甲醛中间体溶解后降温至-2~3℃,然后再与氧化剂混合在20-30℃的条件下反应15-20h;在氧化反应完成之后,进行萃取、浓缩得到氮杂环羧酸中间体。氧化反应在常温下进行即可,反应时间控制在15-20h为宜,以使醛基充分氧化。
s3、氢解反应
将氮杂环羧酸中间体进行氢解,以制备得到氮杂环丁烷-3-羧酸。具体地,氢解过程是将氮杂环羧酸中间体、有机溶剂、催化剂混合后,在1.5-2.5mpa、30-50℃的条件下与氢气反应20-30h。在催化剂存在的条件下,氮杂环羧酸中间体与氢气反应,脱除保护基团,得到氮杂环丁烷-3-羧酸。反应温度控制在30-50℃,反应时间控制在20-30h,更大程度上提升了目标产物的收率。
优选地,催化剂为钯碳催化剂;更优选地,催化剂的用量为氮杂环羧酸中间体质量的3-7%;更优选地,钯碳催化剂中钯的负载量为8-12%。采用钯碳催化剂的催化活性较高,能够加快氢解反应的进行,钯碳催化剂中钯的负载量控制在10%左右为宜,能够降低成本的同时保证反应活性。
优选地,氢解反应完成后,过滤催化剂并进行减压浓缩得到氮杂环丁烷-3-羧酸粗品;将氮杂环丁烷-3-羧酸粗品进行重结晶、过滤,并将滤饼用乙酸乙酯淋洗,再进行干燥。通过初步提纯与再次提纯显著提升目标产物的纯度,去除未反应的原料。
本发明实施例还提供了一种氮杂环丁烷-3-羧酸,应用上述氮杂环丁烷-3-羧酸的制备方法制备而得;优选地,氮杂环丁烷-3-羧酸的纯度为≥98%,更优选为99.0-99.2%。发明人通过优化氮杂环丁烷-3-羧酸的合成路线,不仅缩短了合成路线降低了合成成本,还有利于得到纯度较高的目标产物,适合于推广应用。
以下结合实施例对本发明的特征和性能作进一步的详细描述。
实施例1
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢滴加lihmds317.3克,滴加完毕,0℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在0℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛95.3克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛95.3克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入37.7克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、10%钯碳,升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品35.3克,收率82.8%,gc纯度99.2%。
实施例2
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢滴加lihmds317.3克,滴加完毕,10℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在10℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛86.8克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛86.8克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入34.4克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量10%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的3%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品29.7克,收率69.7%,gc纯度98.8%。
实施例3
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢滴加lihmds317.3克,滴加完毕,5℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在5℃,反应12小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛84.7克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛84.7克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入33.5克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量8%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的7%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品30.3克,收率71.2%,gc纯度98.5%。
实施例4
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500mln,n-二甲基甲酰胺,开启机械搅拌,然后缓慢滴加lihmds317.3克,滴加完毕,0℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在0℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛87.9克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛87.9克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入34.8克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量12%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的7%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品31.8克,收率74.7%,gc纯度99.0%。
实施例5
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢加入叔丁醇钾232.6克,加入完毕,5℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在5℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛76.3克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛76.3克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入34.8克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量10%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的5%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品26.1克,收率61.2%,gc纯度98.6%。
实施例6
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入361.2克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢滴加lihmds264.4克,滴加完毕,3℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在3℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛87.9克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛87.9克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入34.8克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量10%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的5%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品31.1克,收率73.0%,gc纯度99.1%。
实施例7
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢滴加lihmds222.1克,滴加完毕,2℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在2℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛90.0克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛90.0克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入35.6克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量10%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的5%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品31.5克,收率73.9%,gc纯度99.0%。
实施例8
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢滴加lihmds222.1克,滴加完毕,2℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在2℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛93.2克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛93.2克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢滴加67.1克双氧水,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量10%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的5%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品25.5克,收率59.8%,gc纯度98.2%。
实施例9
本实施例所提供的制备氮杂环丁烷-3-羧酸的方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢滴加lihmds317.3克,滴加完毕,1℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在1℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛93.2克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛93.2克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入43.6克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量10%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的5%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品27.0克,收率63.4%,gc纯度98.6%。
实施例10
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体包括以下步骤:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢滴加lihmds317.3克,滴加完毕,1℃搅拌反应2小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在1℃,反应8小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得1-二苯甲基氮杂环丁烷-3-甲醛91.9克。
(2)室温下,三口瓶中依次加入步骤(1)中的1-二苯甲基氮杂环丁烷-3-甲醛91.9克和四氢呋喃溶液、纯化水,开启机械搅拌,降温至0℃,缓慢加入36.4克亚氯酸钠,加入完毕,升温至20~30℃反应15~20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到中间体1-二苯甲基氮杂环丁烷-3-羧酸。在2升氢化反应釜中加入1-二苯甲基氮杂环丁烷-3-羧酸、乙醇、负载量10%钯碳(用量为1-二苯甲基氮杂环丁烷-3-羧酸的5%),升温至40℃,通入氢气,控制压力2mpa反应24小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品32.3克,收率75.9%,gc纯度98.4%。
实施例11
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:甲氧基甲基三苯基氯化膦的用量替换为360.8g,即氮杂环化合物和甲氧基甲基三苯基氯化膦的摩尔比为1:2.5,所得产品氮杂环丁烷-3-羧酸纯品29.7克,收率69.7%,gc纯度98.1%。
实施例12
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:甲氧基甲基三苯基氯化膦的用量替换为577.27g,即氮杂环化合物和甲氧基甲基三苯基氯化膦的摩尔比为1:4,所得产品氮杂环丁烷-3-羧酸纯品24.9克,收率58.5%,gc纯度97.3%。
实施例13
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:甲氧基甲基三苯基氯化膦替换为甲基三苯基溴化膦,所得产品氮杂环丁烷-3-羧酸纯品26.9克,收率63.1%,gc纯度98.3%。
实施例14
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:将原料1-二苯甲基-3-氮杂环丁酮替换为1-苄基氮杂环丁烷-3-酮,具体步骤如下:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢加入氢化钠45.5克,加入完毕,5℃搅拌反应1小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在5℃,反应3小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得氮杂环甲醛中间体59.3g。
(2)室温下,三口瓶中依次加入步骤(1)中的氮杂环甲醛中间体和四氢呋喃溶液、纯化水,开启机械搅拌,降温至3℃,缓慢加入36.4克亚氯酸钠,加入完毕,升温至30℃反应15小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到氮杂环羧酸中间体。在2升氢化反应釜中加入氮杂环羧酸中间体、乙醇、10%钯碳,升温至30℃,通入氢气,控制压力1.5mpa反应20小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品16.2g,收率38.1%,gc纯度96.5%。
实施例15
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:将原料1-二苯甲基-3-氮杂环丁酮替换为1-boc氮杂环丁烷-3-酮,具体如下:
(1)在0℃下,三口瓶中依次加入433.4克甲氧基甲基三苯基氯化膦,500ml四氢呋喃,开启机械搅拌,然后缓慢加入氢化钠57.8克,加入完毕,5℃搅拌反应1小时,缓慢滴加100克1-二苯甲基-3-氮杂环丁酮四氢呋喃溶液,滴加完毕,保持温度在5℃,反应3小时,反应完毕,加入纯化水、乙酸乙酯萃取,减压浓缩得氮杂环甲醛中间体65.6g。
(2)室温下,三口瓶中依次加入步骤(1)中的氮杂环甲醛中间体和四氢呋喃溶液、纯化水,开启机械搅拌,降温至-2℃,缓慢加入13.7克双氧水,加入完毕,升温至20℃反应20小时,反应完毕,加入乙酸乙酯萃取,萃取后经减压浓缩得到氮杂环羧酸中间体。在2升氢化反应釜中加入氮杂环羧酸中间体、乙醇、10%钯碳,升温至50℃,通入氢气,控制压力2.5mpa反应30小时,反应完毕,过滤钯碳,减压浓缩乙醇得到氮杂环丁烷-3-羧酸粗品,经乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯淋洗,干燥,得氮杂环丁烷-3-羧酸纯品13.8g,收率32.4%,gc纯度96.9%。
实施例16
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:将原料1-二苯甲基-3-氮杂环丁酮替换为1-乙酰基氮杂啶-3-酮,所得产品氮杂环丁烷-3-羧酸纯品11.9克,收率27.9%,gc纯度94.8%。
实施例17
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:甲氧基甲基三苯基氯化膦与碱性试剂lihmds的摩尔比为1:0.5,所得产品氮杂环丁烷-3-羧酸纯品1.6克,收率3.7%,gc纯度97.7%。
实施例18
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:甲氧基甲基三苯基氯化膦与碱性试剂lihmds的摩尔比为1:3,所得产品氮杂环丁烷-3-羧酸纯品18.2克,收率42.9%,gc纯度86.4%。
对比例1
本实施例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:甲氧基甲基三苯基氯化膦替换为甲基三苯基溴化膦,原料几乎未参与反应,所得产品氮杂环丁烷-3-羧酸纯品0.5克,收率1.2%,gc纯度79.3%。。
对比例2
本对比例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:氮杂环化合物和甲氧基甲基三苯基氯化膦的反应温度为-5℃,原料几乎不参与反应。
对比例3
本对比例提供一种氮杂环丁烷-3-羧酸的制备方法,具体步骤与实施例10大致相同,不同之处在于:氮杂环化合物和甲氧基甲基三苯基氯化膦的反应温度为20℃,在该反应条件下,原料无法反应完全。
试验例1
将实施例10-18中制备的产品,进行产率和纯度的测试,实施例10-18中的产率依次为:75.9%、69.7%、58.5%、63.1%、38.1%、32.4%、27.9%、3.7%和42.9%;实施例10-18中的纯度依次为:98.4%、98.1%、97.3%、98.3%、96.5%、96.9%、94.8%、97.7%和86.4%。
试验例2
对实施例10中得到产物进行核磁共振氢谱测试,结果如图1:
1hnmr(400mhz,d2o):δ:3.49(m,1h),4.09~4.11(m,4h);
测试结果显示,1~4号的氢归属于δ为4.09~4.11,5号的氢归属于δ为3.49,测试结果正确,所制备的产品为:
实验结果表明,本申请所提供的制备氮杂环丁烷-3-羧酸的制备方法所制备的氮杂环丁烷-3-羧酸具有纯度高等优点,纯度大于98%,最高可以达到99.2%,收率可以达到82.8%。
综上所述,本发明提供的一种用于制备氮杂环丁烷-3-羧酸的氮杂环羧酸中间体,其以1-二苯甲基-3-氮杂环丁酮、1-苄基氮杂环丁烷-3-酮、1-boc氮杂环丁烷-3-酮、1-乙酰基氮杂啶-3-酮等氮杂环化合物为原料,通过与甲氧基甲基三苯基氯化膦进行wittig反应制备得到氮杂环甲醛中间体,再将氮杂环甲醛中间体进行氧化后得到氮杂环羧酸中间体。通过两步反应制备得到氮杂环羧酸中间体,缩短了反应路线,具有原料易得、操作简单安全。
本发明还提供的一种氮杂环丁烷-3-羧酸的制备方法,应用上述制备方法制备氮杂环羧酸中间体,然后经过氢解步骤制备得到氮杂环丁烷-3-羧酸,工艺路线短、原料易得,且操作的安全性高。
以上所描述的实施例是本发明一部分实施例,而不是全部的请对实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!