质粒载体及其应用的制作方法
本发明涉及基因工程
技术领域:
,尤其涉及质粒载体及其应用。
背景技术:
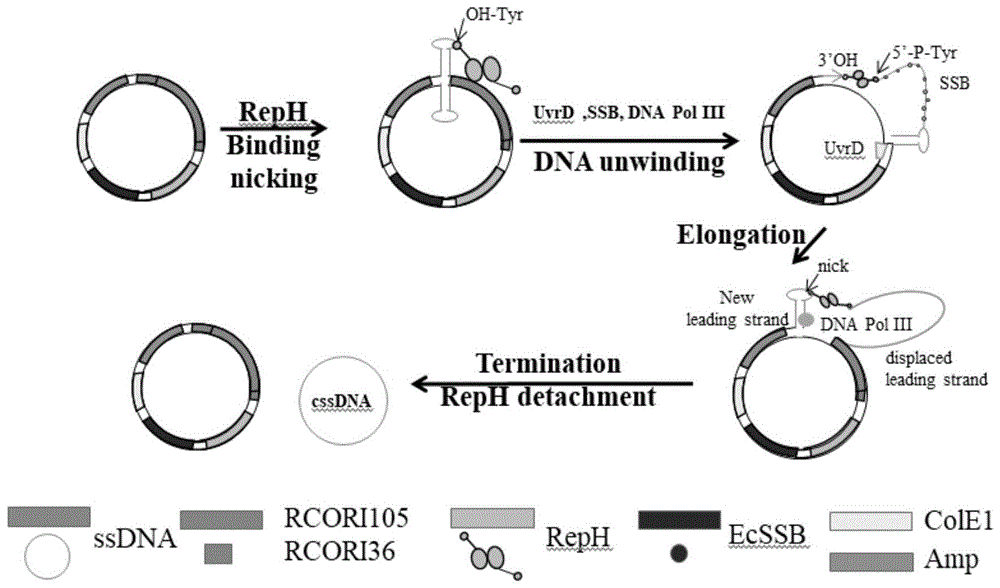
:ssdna在生物学、纳米技术和生物技术中有十分广泛的应用,例如:dna折纸技术等,2006年加州理工学院的rothemund提出了dna折纸术这一全新的dna自组装策略,将dna自组装领域推进到一个新的发展阶段。该方法使用一条长单链dna作为骨架链,上百条序列不同的短链dna作为订书钉链。通过碱基互补作用,订书钉链将骨架链折叠成为所设计的各种形状。在dna折纸术中,所有订书钉链的dna序列不同,从而使整个纳米结构在空间上具有完全的可寻址性。dna折纸技术组装得到的结构可以作为模板,引导其他纳米材料、药物小分子、生物大分子等成分的排列,从而获得光学、电磁学等性能可控的纳米元件、药物载体以及纳米机器人等,使dna折纸术在纳米领域具有非常广泛的应用价值。然而,由于长ssdna的天然来源稀缺,迫切需要一种可以合成序列可控(用户自定义)、成本低廉、纯度高、可大规模生产的较长ssdna的技术。根据所需ssdna的大小、规模和纯度,其生产可能变得异常昂贵或繁重。虽然化学合成的ssdna已在商业上得到广泛应用,但这种dna的长度上限在200(nt)核苷酸之内,而且通常需要额外的处理步骤来去除杂质。可以通过一些生物方法以双链dna(dsdna)为模板经过酶处理、磁珠吸附、滚环扩增、环介导的等温扩增,聚合酶/外切核酸酶/切口酶(pen)反应,引物交换反应(per)级联反应,链置换扩增或不对称pcr方法生产ssdna。例如,pound等使用5’端带生物素修饰的引物进行聚合酶链反应(pcr)扩增,然后使用涂有链霉亲和素的磁珠分离出ssdna。同样是基于pcr的方法,veneziano等使用不对称pcr方法,通过利用一个高度顺向的taq聚合酶,以及在pcr体系中添加数量相差较大的两种引物(正反相引物的摩尔数不同),生产长度出大于15kb的ssdna。但是这些基于pcr的方法通常受到方案的复杂性、可扩展性、回收单链的纯度和成本较高等方面的限制。在体内方法中,x.chen等提出了一种策略,他们首先通过gibsonassembly无缝克隆的方式,将不同来源的特定dna片段与噬菌体的复制起始位点m13origin以及质粒的复制起点pbr322origin进行拼接,组装成双链环状的重组噬菌粒,然后将此重组噬菌粒转化引入包含f因子的大肠杆菌,通过大肠杆菌的繁殖进行大量扩增。经辅助噬菌体侵染后,重组噬菌粒以单链的形式包裹于噬菌体并被分泌到培养液中,经历一系列收集纯化操作后,便可获得相应序列的环状单链dna。通过该方法,他们制备了4种不同长度的单链dna序列,包括长度分别为30000多和20000多及另外两种不同序列的、长度为10000多个碱基的单链环状dna。体内的方法(基于m13噬菌体的方法)在噬菌体内产生ssdna的主要缺点是,其产生的ssdna的序列被限制在噬菌体内稳定繁殖的基因组的序列上,ssdna序列不可控即不能生产用户自定义的ssdna,并且产生的链通常包含大量的载体dna。并且,由于噬菌体具有转染的特性,它会污染其他试剂,为实验室其他成员的实验带来风险,这大大提升了操作的复杂性。因此,迫切需要成本低廉、纯度高、可大规模安全的生产较长ssdna的技术。技术实现要素:有鉴于此,本发明要解决的技术问题在于提供质粒载体及其应用,该质粒可以在细菌体内生产环状单链dna(cssdna),引入表达cas9蛋白的载体,则可产生线性ssdna。而此线性ssdna可用于基因编辑。本发明提供了一种质粒载体,依次包括:单链dna复制起始位点、单链dna复制终止位点和reph表达盒。利用上述质粒载体,可以在表达解旋酶的细胞内生产环状单链dna(cssdna)。该质粒以滚环扩增为基础,其原理如图1,具体而言,reph蛋白特异性地结合到单链dna复制起始位点上,由于reph蛋白的挤压,会导致质粒dna急剧弯曲形成发夹结构,进而促发reph蛋白在位点(g-a)进行切割,形成切口(nick)位点。此时reph蛋白并没有脱落,而是通过其活性位点上的酪氨酸残基与5’磷酸共价连接,同时通过特定的蛋白间相互作用招募解旋酶。解旋酶解开dsdna,直到复制到达单链dna复制终止位点,reph蛋白另一个活性单体在单链dna终止区域的一系列切割/重新连接的作用之后,这个环状的ssdna释放。在解旋酶解开dsdna后,ssdna开始产生,为了防治ssdna降解,可以在表达载体中添加ssb蛋白。这是一种ssdna结合蛋白,其能够包裹在移位的ssdna上,以阻止ssdna降解。一些实施例中,本发明所述的质粒载体中还包括ssb表达盒。本发明所述的质粒载体中,所述单链dna复制起始位点、单链dna复制终止位点之间还包括制备的目的片段。本发明方案对制备的目的片段的长度和核苷酸序列没有特殊要求,本发明实施例中,对长度为2kbp的dna片段进行制备cssdna的验证,结果表明能够成功制备cssdna。一般而言,革兰氏阴性菌较革兰氏阳性菌的代谢更简单,且细胞壁较薄更有利于转化、提取等操作的进行。故而本发明所述cssdna或ssdna的制备优选在革兰氏阴性菌种进行。因此,本发明所述的质粒载体中还包括革兰氏阴性菌的复制起始区。本发明中,所述革兰氏阴性菌为大肠杆菌。经验证,相对于采用中低拷贝数的质粒(例如pet19b),以高拷贝数质粒作为骨架载体更有利于制备过程的进行。一些具体实施例中,用于制备cssdna的所述质粒载体的骨架载体为puc19载体。在以大肠杆菌为重组宿主制备cssdna的实施例中,编码所述ssb蛋白的核酸序列来自大肠杆菌,其genbanksequenceid为cp032667.1。该实施例中,编码所述reph蛋白的核酸序列来自大肠杆菌,其genbanksequenceid为v01277.1。在该实施例中,所述革兰氏阴性菌的复制起始区为cole1,其来自puc19质粒。为了更好的进行筛选和标记,本发明所述的质粒载体中还包括抗性筛选标记。一些实施例中,所述抗性筛选标记为amp标记,所述amp抗性标记的核酸序列来自puc19质粒。在该实施例中,所述质粒载体中各元件的连接顺序为cole1区域、amp抗性标记、单链dna复制起始位点、制备目的片段、单链dna复制终止位点、reph表达盒、ssb表达盒。为了在不含有解旋酶的细胞体内也能够实现cssdna的制备,本发明所述的质粒载体上还包含解旋酶的表达盒。所述编码所述解旋酶的dna序列来自大肠杆菌,具体为uvrd解旋酶。上述质粒载体中,单链dna复制起始位点、单链dna复制终止位点之间的片段即为复制产生cssdna的模板片段。该片段与质粒上的其他区域一样都是双链,其获取可采用现有技术中已知的任何方法进行,本发明实施例中采用pcr的方法对目的片段进行制备。即通过pcr的方式制备与cssdna序列一致的双链dna,其中一条链与需制备的cssdna一致,另一条链与cssdna反向互补。本领域技术人员有能力将人工合成的目的片段通过本领域的常规方法连接入含有单链dna复制起始位点、单链dna复制终止位点和reph表达盒的质粒载体,因此,含有或不含有目的片段的载体,只要其含有单链dna复制起始位点、单链dna复制终止位点和reph表达盒,其都在本发明的保护范围之内。上述质粒载体,能够用于cssdna的制备,即制备环状单链dna。因此,本发明提供了上述质粒载体在cssdna制备中的应用。并且,本发明还提供了携带有上述质粒载体的重组宿主。在本发明实施例中,上述重组宿主的为重组的革兰氏阴性菌。具体的,重组宿主为携带本发明所述质粒载体的大肠杆菌。一些实施例中,所述大肠杆菌为e.colidh5α或dh10β。本发明提供了cssdna的制备方法,其为发酵上述重组宿主。具体为,发酵携带有本发明所述质粒载体的重组宿主并诱导reph蛋白的表达。一些实施例中,发酵的重组宿主携带的质粒载体上包括顺序连接的cole1区域、单链dna复制起始位点、制备目的片段、单链dna复制终止位点、reph表达盒、ssb表达盒。为了实现线性单链dna,即ssdna的制备,可采用酶切的方式将产生的cssdna切割成为线性ssdna,也可采用其他本领域熟知的手段。本发明中,将cas9蛋白引入质粒的受体细胞中从而实现了单链dna的体内切割,缩短了制备的步骤。本发明提供的质粒载体中包括单链dna复制起始位点、单链dna复制终止位点和reph表达盒。为了提高ssdna制备的稳定性,该质粒载体上还包括ssb表达盒。为了实现线性切割,该载体上还包括cas9识别位点。在该质粒载体中,所述cas9的识别位点可为1个,也可为2个或多个。本发明中,所述cas9识别位点的个数为2个,其中cas9识别位点1和cas9识别位点2的序列取决于grna的设计,二者可不同,也可相同。一些实施例中,所述质粒载体中依次包括:单链dna复制起始位点、cas9识别位点1、cas9识别位点2、单链dna复制终止位点和reph表达盒和grna元件。另一些实施例中,所述质粒载体中依次包括:单链dna复制起始位点、cas9识别位点1、cas9识别位点2、单链dna复制终止位点和reph表达盒和ssb表达盒。在该实施例中,所述cas9识别位点1与cas9识别位点2之间还包括制备的目的片段。本发明用于制备ssdna的质粒载体可以在各种含有解旋酶的细胞内实现ssdna的制备,在不含有解旋酶的细胞内,则可在质粒载体上添加解旋酶的表达盒,从而实现ssdna的制备。其中可包括ssb的表达盒也可不包括。基于转化的难易程度以及表达背景较为简洁,本发明所述的质粒载体适宜在格兰仕阴性菌中表达并制备ssdna。一些具体实施例中,ssdna的制备在大肠杆菌中进行。经验证,相对于采用中低拷贝数的质粒(例如pet19b),以高拷贝数质粒作为骨架载体更有利于制备过程的进行。一些具体实施例中,所述用于制备ssdna的质粒载体的骨架载体为puc19载体。在以大肠杆菌为重组宿主制备ssdna的实施例中,编码所述ssb蛋白的核酸序列来自大肠杆菌,其genbanksequenceid为cp032667.1。该实施例中,编码所述reph蛋白的核酸序列来自大肠杆菌,其genbanksequenceid为v01277.1。在该实施例中,所述革兰氏阴性菌的复制起始区为cole1,其来自puc19质粒。为了更好的进行筛选和标记,本发明所述的质粒载体中还包括抗性筛选标记。一些实施例中,所述抗性筛选标记为amp标记,所述amp抗性标记的核酸序列来自puc19质粒。本发明实施例中,所述cas9识别位点1和cas9识别位点2的序列是相同的,具体为:atcaacttcaaactcacaac(seqidno:4)。一些具体实施例中,制备ssdna的质粒载体中各元件的连接顺序为cole1区域、amp抗性标记、单链dna复制起始位点、cas9识别位点1、制备目的片段、cas9识别位点2、单链dna复制终止位点、reph表达盒、ssb表达盒。上述质粒载体中,单链dna复制起始位点、单链dna复制终止位点的片段即为复制产生ssdna的模板片段。该片段与质粒上的其他区域一样都是双链,其获取可采用现有技术中已知的任何方法进行,本发明实施例中采用pcr的方法对目的片段进行制备。即通过pcr的方式制备ssdna的双链dna,其中一条链与需制备的ssdna一致,另一条链与ssdna反向互补。本领域技术人员有能力将人工合成的目的片段通过本领域的常规方法连接入制备ssdna的质粒载体,因此,含有或不含有目的片段的载体,只要其含有单链dna复制起始位点、cas9识别位点1、cas9识别位点2、单链dna复制终止位点和reph表达盒,其都在本发明的保护范围之内。上述质粒载体能够用于ssdna的制备,即制备线性单链dna。因此,本发明本发明提供了上述质粒载体在ssdna制备中的应用。并且,本发明还提供了携带有上述质粒载体的重组宿主。上述重组宿主携带有:cas9表达载体以及上述制备ssdna的质粒载体。在本发明实施例中,上述重组宿主的为重组的革兰氏阴性菌。具体的,重组宿主为携带本发明所述质粒载体的大肠杆菌。一些实施例中,所述大肠杆菌为e.colimg1655。所述cas9表达载体为predcas9。在reph酶表达后,在宿主体内形成cssdna,该cssdna含有cas9识别位点1、左同源臂、donor片段、右同源臂、cas9识别位点2,并含有部分由起始位点和终止位点融合的片段。利用grna指导cas9蛋白进行切割即可获得线性的ssdna片段。本发明提供了ssdna的制备方法,其为发酵携带有:cas9表达载体以及上述制备ssdna的质粒载体的重组宿主,诱导reph蛋白以及cas9蛋白的表达,获得ssdna。一些实施例中,发酵的重组宿主携带的制备ssdna的质粒载体上包括顺序连接的cole1区域、单链dna复制起始位点、cas9识别位点1、制备目的片段、cas9识别位点2、单链dna复制终止位点、reph表达盒、ssb表达盒。本发明制备的ssdna片段含有制备目的片段以及位于两端的cas9识别位点被切割后的残余片段。该片段可用于诊断、治疗、基因合成及测序、dna折纸技术和dna信息存储等多个领域。本发明还提供了一种质粒载体,其中包括单链dna复制起始位点、单链dna复制终止位点和reph表达盒。为了制备获得的ssdna能够顺利脱离并与目标区域结合,此载体中不含有ssb表达盒。为了实现ssdna的线性化,该载体上还包括cas9识别位点。所述cas9识别位点的数量为2个,位于单链dna复制起始位点、单链dna复制终止位点之间。本发明中,所述cas9识别位点1和cas9识别位点2的序列取决于sgrna的设计,二者可不同,也可为序列相同的两个位点。该载体上还包括grna元件。所述grna元件包括启动子、grna序列以及支架。所述grna序列根据编辑的靶基因设计。该载体上还可以包括基因编辑目的片段。所述基因编辑目的片段位于所述cas9识别位点1和cas9识别位点2之间。上述质粒载体中,所述基因编辑的目的片段包括靶基因的左同源臂、donor片段和右同源臂。本发明中,所述donor片段在基因的野生型序列基础上删除、添加或替换一个至多个核苷酸。所述基因编辑目的片段或grna元件与质粒上的其他区域一样都是双链,可采用现有技术中已知的任何方法对其进行制备,本发明实施例中采用pcr的方法对目的片段进行制备。本领域技术人员有能力将人工合成的基因编辑目的片段或grna元件通过本领域的常规方法连接入本发明所述的,含有单链dna复制起始位点、单链dna复制终止位点和reph表达盒的质粒载体,因此,含有或不含有目的片段或grna的载体,只要其含有单链dna复制起始位点、单链dna复制终止位点和reph表达盒,其都在本发明的保护范围之内。一些实施例中,所述质粒载体中依次包括:单链dna复制起始位点、cas9识别位点1、基因编辑目的片段、cas9识别位点2、单链dna复制终止位点和reph表达盒和grna元件。本发明用于基因编辑的质粒载体可以在各种含有解旋酶的细胞内实现基因的编辑。在不含有解旋酶的细胞内,则可在质粒载体上添加解旋酶的表达盒,从而产生解旋酶促进ssdna的产生,从而用于基因编辑。在本本发明的实施例中,利用本发明所述的载体在大肠杆菌体内实现了基因编辑,因大肠杆菌自身可以产生解旋酶uvrd,因此本发明实施例中制备的载体中并没有包含解旋酶表达盒。本发明提供的该质粒载体中,还包括复制起始区。上述复制起始区针对带编辑的生物体进行设计。经验证,相对于采用中低拷贝数的质粒(例如pet19b),以高拷贝数质粒作为骨架载体更有利于ssdna的产生,从而产生更高拷贝数的片段来进行基因编辑。一些具体实施例中,所述用于基因编辑的质粒载体的骨架载体为puc19载体。在本发明实施例中,编码所述reph蛋白的核酸序列来自革兰性阳性菌质粒pc194,其genbanksequenceid为v01277.1。在该实施例中针对大肠杆菌进行基因编辑,因此,上述质粒来自革兰氏阴性菌,具体的复制起始区为cole1,其来自puc19质粒。为了更好的进行筛选和标记,本发明所述的质粒载体中还包括抗性筛选标记。一些实施例中,所述抗性筛选标记为amp标记,所述amp抗性标记的核酸序列来自puc19质粒。本发明实施例中,所述cas9识别位点1和cas9识别位点2的序列是相同的,具体为:atcaacttcaaactcacaac。所述grna元件中,所述启动子为pj23119,序列为ttgacagctagctcagtcctaggtataatactagt(seqidno:5),所述支架序列为gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtg(seqidno:6)。一些具体实施例中,制备ssdna的质粒载体中各元件的连接顺序为cole1区域、amp抗性标记、单链dna复制起始位点、cas9识别位点1、左同源臂、donor片段、右同源臂、cas9识别位点2、单链dna复制终止位点、reph表达盒、grna元件。在携带本发明所述的质粒载体的宿主体内引入的含有cas9的表达载体表达出cas9蛋白,其识别上下游的cas9识别位点并对其进行切割,从而获得包含左同源臂、donor片段和右同源臂的线性ssdna片段,该片段的两端还含有cas9识别位点被切割后的残余片段。该片段与基因组目的区域结合,从而在生物体内引入编辑后的片段。因此,本发明提供了上述质粒载体在基因编辑中的应用。在宿主体内引入的cas9表达载体和λred表达系统,以进行基因编辑。cas9式组合型表达启动子,λred需要iptg诱导表达,还需要包括αβγ片段。在本发明实施例中,针对大肠杆菌lacz基因进行编辑,grna序列为gttgtgagtttgaagttgat(seqidno:11)。本发明还提供了一种基因编辑方法,其将本发明所述的质粒载体转化入携带含有cas9片段和αβγ片段的质粒载体的受体细胞。转入受体细胞后,还包括传代的步骤。在传代后,还包括诱导reph蛋白表达的步骤。本发明所述基因编辑方法使用的受体细胞包括细菌、真菌、植物或动物。在本发明实施例中,采用大肠杆菌验证基因编辑的效果。一些具体实施例中,所述含有cas9片段和αβγ片段的质粒载体为predcas9。在本发明涉及的质粒载体中,所述reph表达盒中,包括:启动子、编码reph的核酸和终止子。一些实施例中,所述启动子为prhabad,其核苷酸序列为tgtgaacatcatcacgttcatctttccctggttgccaatggcccattttcctgtcagtaacgagaaggtcgcgaattcaggcgctttttagactggtcgta(seqidno:7),所述终止子为t1terminator,其核苷酸序列为ccggcttatcggtcagtttcacctgatttacgtaaaaacccgcttcggcgggtttttgcttttggaggggcagaaagatgaatgactgtccacgacgctatacccaaaagaaa(seqidno:8)。在本发明涉及的质粒载体中,所述ssb表达盒中,包括:启动子、编码ssb的核酸和终止子。一些实施例中,所述启动子为placiq,其核苷酸序列为tggtgcaaaacctttcgcggtatggcatgatagcgcccggaagagagtcaattcagg(seqidno:9),所述终止子为t500,其核苷酸序列为acagaaaagcccgcctttcggcgggctttg(seqidno:10)。本发明还对质粒载体中的所述单链dna复制起始位点、单链dna复制终止位点的序列进行了筛选和优化,在一些实施例中,所述单链dna复制起始位点的序列如seqidno:1所示,所述单链dna复制终止位点的序列如seqidno:2或3所示。本发明通过reph蛋白的特异性切口和连接作用,即可产生目标cssdna。工作主要包含质粒结构的设计及cssdna的验证。本发明利用滚环复制机理,在已经作为工程菌广泛使用的大肠杆菌体内生产合成环状ssdna。该发明已成功的证明,在大肠杆菌体内,可以高效快速的产生环状ssdna,并利用cas9蛋白在无前间区序列邻近基序(pam位点),只有指导rna(sgrna)存在的条件下,可以特异性切割单链dna的性能,将体内产生的环状ssdna有效的切割为线性ssdna并用于基因编辑。附图说明图1示cssdna产生原理;图2示实施例1中以pcr确认cssdna存在,其中泳道m为marker,泳道1示以未经酶切的质粒提取物为模板,泳道2示以经过酶切的质粒提取物为模板;图3示cssdna的pcr产物测序结果;图4示比较purs55-2k与purs53-2k的环状ssdna产量(即比较65和36作为终止子的终止能力),其中,泳道m为marker;泳道1为purs53-2k(冻融细胞破碎处理);泳道2为purs55-2k(冻融细胞破碎处理);图5示荧光探针结合实验,其中,泳道m为marker;泳道1为对照,pcsh1-dab-mb降解后的环状ssdna+probe1;泳道2为purs55-2k冻融细胞破碎处理+probe1;泳道3为purs55-2k冻融细胞破碎处理+probe2;泳道4为purs55-2k冻融细胞破碎处理+probe3;图6示对大肠杆菌体内的lacz基因进行编辑原理示意图;图7示平板白斑情况;其中7-a示第1代平板与第10代平板对比;7-b示其中一个第一代平板,7-c示第一代平板中一个白色菌落的局部放大;图8示突变序列测序与未突变序列比对。具体实施方式本发明提供了质粒载体及其应用,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本
发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本发明采用的试材皆为普通市售品,皆可于市场购得。下面结合实施例,进一步阐述本发明:实施例1cssdna的制备1、质粒构建(purs53-2k):通过一系列的同源重组将表1中的目的基因连接到载体puc19上;表1目的基因来源与序列2、构建完成并测序正确的质粒(记为purs53-2k)转化到大肠杆菌感受态dh10β中(图1示cssdna产生过程);3、复苏培养1h后,取30ul接种到3mllb培养基中进行培养(培养时间<12h),1:100稀释接种到新鲜lb培养基中,培养至od=0.5左右,加入鼠李糖(终浓度1mm)诱导表达reph进行cssdna的生产,养菌到od≈1时,回收菌体后,破碎细胞并提取质粒。4、对提取后的质粒(含有原质粒及表达出的环状ssdna)进行酶切和pcr验证,确认是否有ssdna产生。4.1以bamhi和hindiii对提取获得的质粒进行酶切,bamhi位于质粒reph基因上(reph终止子之前),可以切断质粒;hindiii位于质粒ssdna上(36bp终止位点之前),可以同时切断质粒及产生的环状产物(因为这里的目的是验证产生的环状dna是否为单链),酶切后只有cssdna保持完整,能够获得扩增产物。酶切体系表2所示。表2酶切反应体系组分体系质粒dna5μl(1μg)10×buffer5μlbamhi1μlhindiii1μlddh2o到50μl4.2pcr验证cssdna,分别以质粒dna和酶切后的产物作为模板进行pcr验证。扩增正向引物序列为:gcagtcaggccacaattac、扩增的反向引物为:aactgtcctctgcaggtg。反应体系如表3所示。表3pcr反应体系pcr程序如表4所示:表4pcr反应程序结果如图2所示,其中泳道1为由发酵后菌体中提取获得的质粒直接进行pcr的产物电泳结果,泳道2为经酶切的质粒进行pcr所得产物的电泳结果,如图2所示,泳道1在4871bp和1893bp处有存在扩增条带,说明4871bp为质粒为模板的扩增产物;1893bp为环状ssdna为模板的扩增产物,所用pcr引物为:pcr-2k-f:gcagtcaggccacaattac;pcr-2k-r:aactgtcctctgcaggtg,泳道2在1893bp处存在扩增条代,说明酶切完全,质粒为模板的条带产物基本消失,所用同为pcr引物为:pcr-2k-f:gcagtcaggccacaattac;pcr-2k-r:aactgtcctctgcaggtg。将泳道2中获得的,大小约为1893bp的条带进行切胶回收并测序后,证明确为连接上的cssdna的pcr产物,测序结果如图3所示。如图所示:测序的位置位于环状单链dna连接区域,由于连接后序列与连接前序列不同,且如果没有连接为环状,是不能pcr出1893bp的产物条带的,因此,能够证明所获得的产物是环状结构。实施例2cssdna的制备1、质粒构建(purs55-2k):通过一系列的同源重组将表5中的目的基因连接到载体puc19上;表5目的基因来源与序列名称序列ssb启动子placiqseqidno:9ecssbgenbanksequenceid:cp032667.1ssb终止子t500seqidno:10reph启动子prhabadseqidno:7rephgenbanksequenceid:v01277.1reph终止子t1terminatorseqidno:8单链dna复制起始位点(rcori105)seqidno:1单链dna复制终止位点(rcori65)seqidno:3cole1(ori)puc19质粒本身的复制区域amppuc19质粒本身的氨苄抗性基因制备的目的片段seqidno:122、构建完成并测序正确的质粒(记为purs55-2k)转化到大肠杆菌感受态dh10β中(图1示cssdna产生过程);3、复苏培养1h后,取30μl接种到3mllb培养基中进行培养(培养时间<12h),1:100稀释接种到新鲜lb培养基中,培养至od=0.5左右,加入鼠李糖(终浓度1mm)诱导表达reph进行cssdna的生产,养菌到od≈1时,回收菌体后,破碎细胞并提取质粒。ssdna产量比较1、对实施例1和实施例2表达环状单链dna后的细胞用冻融的方法进行细胞破碎后,pcr验证,比较ssdna产量pcr验证cssdna,分别以质粒dna和酶切后的产物作为模板进行pcr验证。扩增正向引物序列为pcr-2k-f1:gctttcacttttatcctgtgc、扩增的反向引物为pcr-2k-r1:tgggggatgtggagaaag。反应体系如表6所示。表6pcr反应体系组分体系模板1μl10×buffer5μleasytaq1μldntps4μlpcr-2k-f11μlpcr-2k-r11μlddh2o到50μlpcr程序如表7所示:表7pcr反应程序目的条带长度为1978bp为环状ssdna为模板的扩增产物,所用pcr引物为:pcr-2k-f1:gctttcacttttatcctgtgc;pcr-2k-r1:tgggggatgtggagaaag对应的图4说明,以65bp作为终止时,在相同条件的pcr后,从pcr产物可以看出,转化purs55-2k质粒的细胞中环状ssdna产量明显高于转化purs53-2k质粒的细胞。cssdna检测对实施例2制备的表达环状单链dna后的细胞用冻融的方法进行细胞破碎后,利用荧光探针与单链dna的互补作用,进一步验证细胞中存在cssdna(图5)。表8探针序列探针名称序列probe1fam-gattattccagggtaattgtggcctgactgprobe2rox-agtgggttacatcgaactggatctcaacagprobe3rox-cagtcaggccacaattaccctggaataatc注:探针1:与cssdna互补,fam修饰;探针2:与amp互补,该互补连为质粒后随链,rox修饰;探针3:与探针1互补;rox修饰。实验以现有技术中的pcsh1-dab-mb载体作为对照,pcshi-dab-mb通过切口酶humi和外切酶exoiii降解得到(序列如seqidno:13),探针采用探针1。表9探针结合体系组分体系细胞破碎物5μl10×easytaqbuffer3μl探针1/2/31μlddh2o到30μl通过以上实验,证明本发明提供的载体能够使大肠杆菌生产cssdna,而由于该反应只要reph存在就可进行,并且细胞可传代,因此,该方法能够在大肠杆菌体内连续生产cssdna。根据结果显示,以rcori65为单链dna复制终止位点能够取得更好的制备效果,因此之后采用rcori65为单链dna复制终止位点进行ssdna的制备以及基因编辑。实施例3ssdna的制备以及基因编辑在本实施例中,将验证在cas9蛋白的作用下,将体内产生的环状ssdna有效的切割为线性ssdna并用于基因编辑。1、质粒构建:通过一系列的同源重组将表8中的目的基因连接到载体puc19上,将该载体记为pur55-grd,利用pur55-grd(有rcori105/65ori,reph,grna,donor结构)和predcas9质粒进行基因编辑,同时,设置不含有grna的对照组,该组质粒中连接有rcori105/65ori,reph,donor片段,但不含有grna元件,记为pur55-d;并设置仅连接donor结构的对照组,该组质粒中仅连接有donor结构,不含有rcori105/65ori,reph,记为puc57-d。表10目的基因来源与序列注所述目的片段中包括同源左臂、donor片段、同源右臂,相对于大肠杆菌中的野生型lacz基因,donor片段突变了其中的11bp,产生4个连续的终止子(tag),能够导致lacz基因失活。2、在大肠杆菌中转化入表达cas9蛋白以及含有αβγ元件的载体predcas9,之后将转化后的大肠杆菌制成感受态;3、将步骤1中构建完成并测序正确的质粒转化到步骤2制成的大肠杆菌感受态中;4、复苏培养1h后,取适量菌液(10-6次方稀释后的100μl)涂布平板,挑取单菌落,过夜培养,1:100稀释接种到新鲜lb培养基中,诱导表达reph进行cssdna的生产,由于菌体内还表达cas9蛋白,因此cssdna产生后cas9蛋白通过grna识别cssdna上的识别位点,从而将cssdna切割成为线性的ssdna,该ssdna在基因复制的过程中充当模板,从而对大肠杆菌体内的lacz基因进行编辑(如图6)。涂板培养后,通过蓝白斑比率和测序,确定编辑效率。实验表明,在实验组中,第一代就出现白斑(图7),且通过测序,验证编辑成功,成功突变11个碱基,使序列产生4个连续的终止密码子(图8)。而对照组pur55-d(与实验组对比,结构中不含有grna)和puc57-d(与实验组对比,结构中只含有donordna片段)均未看到白斑(即编辑成功的菌落)。通过传代培养,确认白斑比例会随着传代次数的增加而增加,传代到第十代后,实验组的编辑效率在41.45%。对照组pur55-d在传菌至第十代后,出现白斑,编辑效率为0.73%.(所述编辑效率=平板上白斑数/平板菌落总数×100%,实验至少重复3次)以上仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。序列表<110>天津大学<120>质粒载体及其应用<130>mp1918122<160>13<170>siposequencelisting1.0<210>1<211>105<212>dna<213>人工序列(artificialsequence)<400>1taaaggatttgagcgtagcgaaaaatccttttctttcttatcttgataataagggtaact60attgccggcgaggctagttacccttaagttattggtatgactggt105<210>2<211>36<212>dna<213>人工序列(artificialsequence)<400>2ttctttcttatcttgataataagggtaactattgcc36<210>3<211>65<212>dna<213>人工序列(artificialsequence)<400>3aaaaatccttttctttcttatcttgataataagggtaactattgccggcgaggctagtta60ccctt65<210>4<211>20<212>dna<213>人工序列(artificialsequence)<400>4atcaacttcaaactcacaac20<210>5<211>35<212>dna<213>人工序列(artificialsequence)<400>5ttgacagctagctcagtcctaggtataatactagt35<210>6<211>75<212>dna<213>人工序列(artificialsequence)<400>6gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagt60ggcaccgagtcggtg75<210>7<211>101<212>dna<213>人工序列(artificialsequence)<400>7tgtgaacatcatcacgttcatctttccctggttgccaatggcccattttcctgtcagtaa60cgagaaggtcgcgaattcaggcgctttttagactggtcgta101<210>8<211>113<212>dna<213>人工序列(artificialsequence)<400>8ccggcttatcggtcagtttcacctgatttacgtaaaaacccgcttcggcgggtttttgct60tttggaggggcagaaagatgaatgactgtccacgacgctatacccaaaagaaa113<210>9<211>57<212>dna<213>人工序列(artificialsequence)<400>9tggtgcaaaacctttcgcggtatggcatgatagcgcccggaagagagtcaattcagg57<210>10<211>30<212>dna<213>人工序列(artificialsequence)<400>10acagaaaagcccgcctttcggcgggctttg30<210>11<211>20<212>dna<213>人工序列(artificialsequence)<400>11gttgtgagtttgaagttgat20<210>12<211>1905<212>dna<213>人工序列(artificialsequence)<400>12agtttggtggtgttgatttgcccttaacctcacataccacctcgaggagctctcaagtcc60tcagctgcaagaatatctgaatgtcttttggagtgttagagtcctctgtgtcttagaaat120tttgaaaagaaaaacaaatctcaatattaatgttgattagtttctctgagccaattgggg180aaaataaacgtccttcacctcaaaggtttaagtgacaccgaagggtagccaccagtgtct240cggccactgaagcctcatgcatgctctcactaccagtttgatttgcagccccatagttgt300gttgtactaaatattctttcctctggccttgtccagtgaacacggttcacatggctaaca360ccacttcttgagatgcgagcaccatgcaaagctgagaacggattgggttttgtgaccatt420gtgcctcctcctcacctgagaggcccatttttcctggttgattcattaagtgtattggtg480ctgtcagtcgcctctggacaattgaaatgacaagtggctgttgattcataaagaaaatga540aggctttagatgtgaaaccctcgttttctcttgtccttctcttaggtgaaagattttatt600tttttcaaaaggctacatactggtatcccagcaggtgtagtgtgagaactggcatatgtt660aggctatggtgtcagtgtggatgggcaattcttcaagatggaaaaccaagtctcactgag720ttgctggagccacactgacctttctccacatcccccaccatgggctttcacttttatcct780gtgcttgaatttttttcacatacaaattctttatacacacacacagacacacacacacat840atctcactctgtcaatgcagtggctgaatcatgggtcactgcatcttcaaattcttaggc900tccagtgatgctttcaaatcagcctctcaagtagctgggactacaggcatgcaaagctac960acccagacaatttttaaatatttttctagagactgagcctacttatgttgctcagactcg1020tcttgcactcctgggatcaaacgataatcccaccttgaccacccaaagtgtttagattac1080aggtgtgagctagcactctcagcaaaaatatattttaaagaaccgttacaaccaaattat1140gagttatcattatgccactgccctccaccctgggcaccagaacaagaccttgtatccaaa1200aactaagcaaaactaaacaagaacaaaaaaaaaaaaacttataaataaattaaactttga1260agattgtgtcatctgtgtccttccctgccctccaagctatcaatgttaaatataatggtt1320attgagaaaatggttagatattattaagaaatttctatatatcttccagctgagaatagg1380tattctgttgtggcccaaatattttctcaccgctaccttcagggtctaaactagcaaatc1440aggacacctgcagaggacagttggccgttttcaaatagaaagagaaatacccccgttcat1500gagagtaatccagtgattttcaaaaagacaagtcagactgacatgcagcgcagtcaggcc1560acaattaccctggaataatcacttcacacagaatggttgaggagactttctaagatgagc1620aaatttgggcagcataatccttgcttatttattcccagcccccactgcccgcctgattcc1680taatggctaccctacaatgtggtcagcagtgggatgtagcgtggtgagagaggggctcag1740ggacgggatgaaggtctttcctgcattatcaaaatgcaggttaaaaagttgttaaaaaga1800tgtccaaatgttctaattcctactgttaaatagctgctaagatgcattatacaacagacc1860caggtaagggagctcggtaccgtcgacctgcaggcatgcaagctt1905<210>13<211>2178<212>dna<213>人工序列(artificialsequence)<400>13cagtgagcgcaacgcaattaatgtgagttagctcactcattaggcaccccaggctttaca60ctttatgcttccggctcgtatgttgtgtggaattgtgagcggataacaatttcacacagg120aaacagctctgttgagatccagttcgatgtaacccacttctgctctgatgccgcatagca180gtcaggccacaattaccctggaataatcgaaagggcctcgtgatacgcctatttttatag240gttaatgtcatgataataatggtttcttattaacagagtaacctcctcaaagtaatgagc300ctaacgctcagcaattcccacttagacgtcaggtggcacttttcggggaaatgtgcgcgg360aacccctatttgtttatttttctaaatacattcaaatatgtatccgctcatgagacaata420accctgataaatgcttcaataatattgaaaaaggaagagtatgagtattcaacatttccg480tgtcgcccttattcccttttttgcggcattttgccttcctgtttttgctcacccagaaac540gctggtgaaagtaaaagatgctgaagatcagttgggtgcacgagtgggttacatcgaact600ggatctcaacagcggtaagatccttgagagttttcgccccgaagaacgttttccaatgat660gagcacttttaaagttctgctatgtggcgcggtattatcccgtattgacgccgggcaaga720gcaactcggtcgccgcatacactattctcagaatgacttggttgagtactcaccagtcac780agaaaagcatcttacggatggcatgacagtaagagaattatgcagtgctgccataaccat840gagtgataacactgcggccaacttacttctgacaacgatcggaggaccgaaggagctaac900cgcttttttgcacaacatgggggatcatgtaactcgccttgatcgttgggaaccggagct960gaatgaagccataccaaacgacgagcgtgacaccacgatgcctgtagcaatggcaacaac1020gttgcgcaaactattaactggcgaactacttactctagcttcccggcaacaattaataga1080ctggatggaggcggataaagttgcaggaccacttctgcgctcggcccttccggctggctg1140gtttattgctgataaatctggagccggtgagcgtgggtctcgcggtatcattgcagcact1200ggggccagatggtaagccctcccgtatcgtagttatctacacgacggggagtcaggcaac1260tatggatgaacgaaatagacagatcgctgagataggtgcctcactgattaagcattggta1320actgtcagaccaagtttactcatatatactttagattgatttaaaacttcatttttaatt1380taaaaggatctaggtgaagatcctttttgataatctcatgaccaaaatcccttaacgtga1440gttttcgttccactgagcgtcagaccccgtagaaaagatcaaaggatcttcttgagatcc1500tttttttctgcgcgtaatctgctgcttgcaaacaaaaaaaccaccgctaccagcggtggt1560ttgtttgccggatcaagagctaccaactctttttccgaaggtaactggcttcagcagagc1620gcagataccaaatactgttcttctagtgtagccgtagttaggccaccacttcaagaactc1680tgtagcaccgcctacatacctcgctctgctaatcctgttaccagtggctgctgccagtgg1740cgataagtcgtgtcttaccgggttggactcaagacgatagttaccggataaggcgcagcg1800gtcgggctgaacggggggttcgtgcacacagcccagcttggagcgaacgacctacaccga1860actgagatacctacagcgtgagctatgagaaagcgccacgcttcccgaagggagaaaggc1920ggacaggtatccggtaagcggcagggtcggaacaggagagcgcacgagggagcttccagg1980gggaaacgcctggtatctttatagtcctgtcgggtttcgccacctctgacttgagcgtcg2040atttttgtgatgctcgtcaggggggcggagcctatggaaaaacgccagcaacgcggcctt2100tttacggttcctggccttttgctggccttttgctcacatgttctttcctgcgttatcccc2160tgattctgtggataaccg2178当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1